https://www.youtube.com/watch?v=ogM3S1jimIY
LATAA ITSELLESI HETI TÄMÄ VIDEO!
Tässä dokumenttielokuvassa paljastetaan miten pieni itsekäs ahne psykopaattijoukko on ohjannut maailman menoa nykyhistorian aikana ja miten se on yhä tänäpäivänä poliittisen vallan takana, miten se ohjaa maailmaa ja mikä on sen paha suunnitelma ihmiskunnalle.
KATSO TÄMÄ HETI, NIIN TIEDÄT ENEMMÄN.


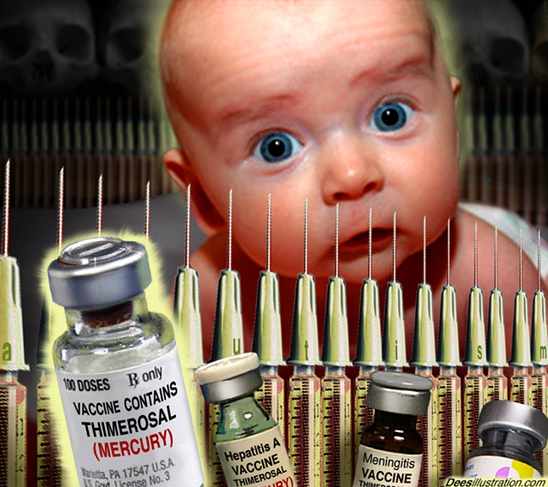
Kaiken väärän tiedon, jota levitetään tappavista viruksista joukkotiedotusvälineiden kautta, tarkoituksena on pitää ihmiskunta kontrolloituna ja tietämättömyydessä.

Ebola on USA:n hallituksen vuonna 2010 patentoima virus ja sillä on omistamansa patentin omistusoikeus tämän patentin kautta kaikkiin Ebola-viruksiin. US Centers for Disease Control (CDC) omistaa Ebola patentin.

http://www.youtube.com/watch?v=6bPDBND2jL4
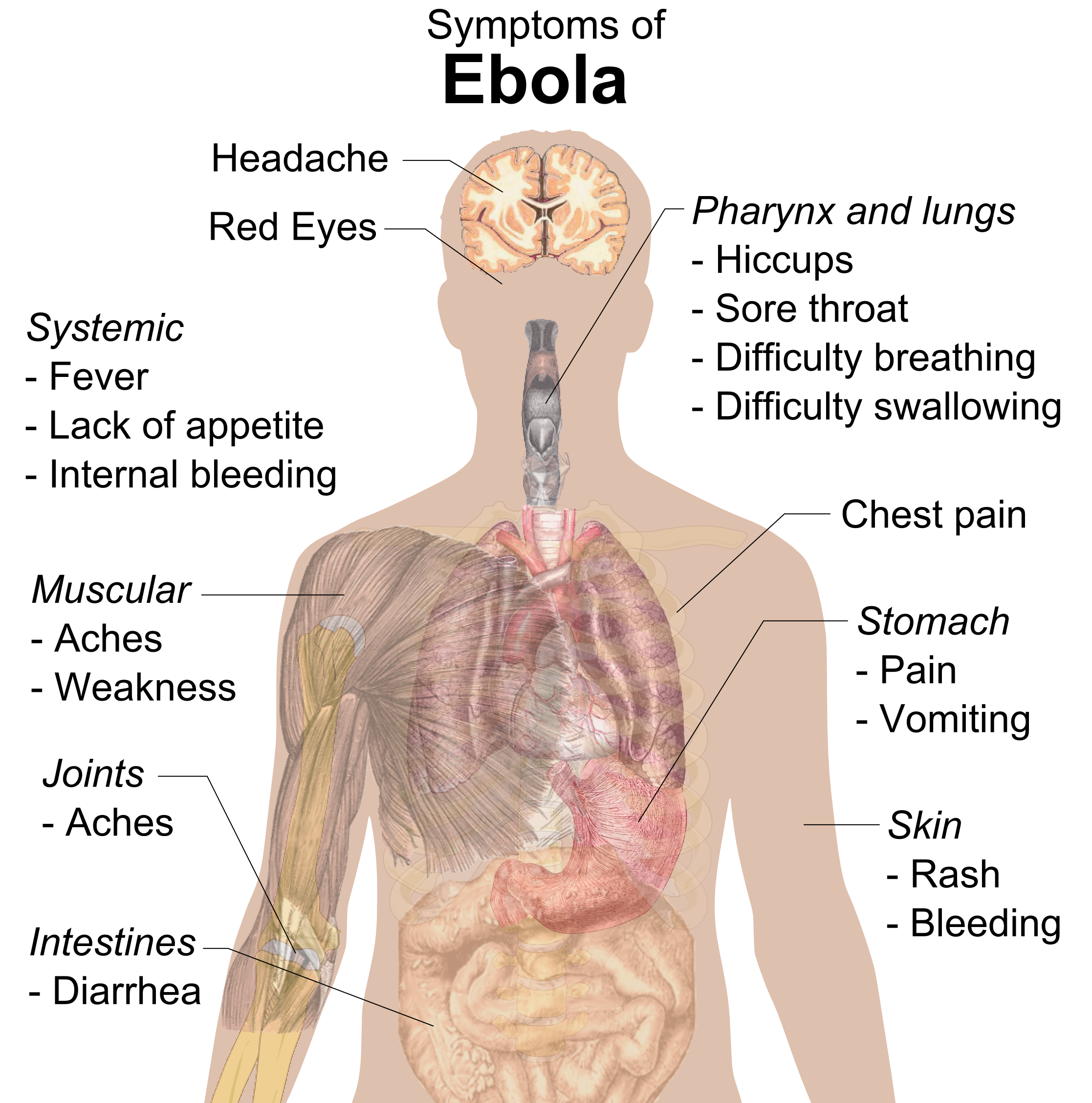
AIDS ja Ebola virukset on biologisesti kehitetty USA:n hallituksen laboratorioissa. Ebola patenttiin on USA:lla monopoli, jotta se voi yksinomaan "hyötyä" tästä "keksinnöstä" ja estää muita "hyödyntämästä" tätä "keksintöään". USA saa tästä "EBOLA-keksinnöstä" itselleen kaiken "voiton" ja "hyödyn".
http://www.naturalnews.com/046290_Ebola_patent_vaccines_profit_motive.html

Tohtori Cyril Broderick, liberialainen tiedemies ja entinen kasvitautiopin professori Liberian yliopiston maa- ja metsätalousministeriöstä kertoo lännen, erityisesti Yhdysvaltain, olevan vastuussa Ebola -epidemiasta Länsi-Afrikassa.

Liberialainen tiedemies:
Yhdysvallat on vastuussa
Ebola-epidemiasta
Tohtori Broderick väittää seuraavaa yksinoikeudella julkaistussa Daily Observer jehden artikkelissa Liberiassa:
Yhdysvaltain puolustusministeriö (DoD) rahoittaa Ebola kokeita ihmisillä, kokeita, jotka alkoivat vain viikkoja ennen Ebola -epidemiaa Guineassa ja Sierra Leonessa.
Raportit kertovat, että DoD teki sopimuksen arvoltaan 140 000 000 dollaria Tekmiran, kanadalaisen lääkeyhtiön kanssa, tehdä tätä "Ebola tutkimusta".

Raportit kertovat, että DoD teki sopimuksen arvoltaan 140 000 000 dollaria Tekmiran, kanadalaisen lääkeyhtiön kanssa, tehdä tätä "Ebola tutkimusta".
Tämä "tutkimustyö" piti sisällään terveiden ihmisten suonensisäiset injektiot tappavalla Ebola-viruksella, joka alkoi vuonna 2014 tammikuussa, siis vain hieman ennen kuin Ebola -epidemia julistettiin Länsi-Afrikassa maaliskuussa.



Yhdysvaltain puolustusministeriö (DOD) ja muut länsimaat ovat suoraan vastuussa tartuttaessaan afrikkalaiset Ebola-viruksella!

Dr. Broderick väittää, että Yhdysvaltain hallituksella on tutkimuslaboratorio Sierra Leonen kaupungissa nimeltään Kenema, joka tutkii, mitä hän kutsuu "viruskuume bioterrorismiksi".
Kaupungissa on myös "Ebola-epidemian keskus” Länsi-Afrikassa.
Samalla tavalla rokotuskampanjoin USA:a kontrolloiman porukan vaikutuksesta levitettiin Belgian Kongosta alkaen hiv-virusta ja Manhattanilla homoseksuaalien keskuuteen, jotta se sai tapettua ihmisiä.

Hän sanoo edelleen, että "on kiire alkaa suojella vähemmän varakkaiden ja köyhempien maiden, erityisesti Afrikan kansalaisia, joiden maat eivät ole tieteellisesti ja teollisesti varustettu kuten Yhdysvallat ja useimmat länsimaat, joissa on useimmat strategisesti suunniteltujen biologisten aseiden, kuten virusten tai GMO -bakteerien laboratorioista.

Hän myös kysyy tärkeän kysymyksen, kun hän sanoo: on erittäin huolestuttavaa, että Yhdysvaltojen hallitus on toiminut verenvuotokuume bioterrorismin tutkimuslaboratoriossa Sierra Leonessa.
Onko muita?
Onko muita?
Mitä valtamedia ei halua sinun tietävän Ebolasta?
Guatemalan kuppakokeet: Kuinka USA: n johtama ryhmä suoritti ihmiskokeita Keski-Amerikassa
Herra Broderickin väitteet näyttävät olevan totta. Loppujen lopuksi Yhdysvaltain hallitus on kokeillut tappavia tauteja ihmisiin pitkään, historia kertoo tämän. Yksi esimerkki on Guatemala.
1946 - 1948 aikana Yhdysvaltain hallitus presidentti Harry S. Truman (mies, joka päätti atomipommien tarpeettomasta pudotuksesta Japaniin kun maa oli jo sitä ennen ilmoittanut halukkuutensa rauhaan /oma lisäys) yhteistyössä Guatemalan presidentti Juan José Arévalon ja hänen terveysviranomaistensa kanssa tarkoituksella tartutti yli 1 500 sotilasta, prostituoitua, vankeja ja mielenterveyspotilaita kuppaan ja muihin sukupuolitauteihin, kuten tippuriin ja bakteeri infektioihin, yhteensä yli 5 500 Guatemalan kansalaiseen, jotka osallistuivat kokeisiin.
Epäeettistä tutkimusta ei ole ilmoitettu julkisesti kuin vasta äskettäin, kun presidentti Obamaklaaneineen pyysi anteeksi Guatemalan hallitukselta ja kansalta sekä lupasi olla koskaan toistamatta menneisyyden virheitä - aikana, jolloin ei ollut harvinaista, että lääkärit tekivät kokeita potilaille ilman heidän suostumustaan.
Tutkimusta Guatemalassa johti tohtori John Cutler, USA terveydenhuollon lääkäri, joka osallistui myös kiisteltyihin Tuskegeen kuppa kokeisiin, jotka alkoivat 1930-luvulla. Komissio sanoi 5 500 guatemalalaisen olleen mukana kaikissa tutkimuksissa, jotka tehtiin 1946-48. Näistä noin 1300 sai tarkoituksella kuppatartunnan, tippurin tai muun sukupuolitaudin.
Yhdysvaltain hallitus on syyllistynyt useisiin lääketieteellisiin ihmiskokeisiin ei vain Guatemalassa vaan myös muissa maissa sekä omalla alueellaan. Kuten Boston Globe raportti kertoo, Tuskegeen kuppatutkimus tapahtui vuosina 1932 ja 1972.
Yhdysvaltain valtion virastoilla on pitkä historia suorittamistaan biologisen sodankäynnin tutkimuksista Liberiassa ja Sierra Leonessa. Miksi Obaman hallinto lähettää joukkoja Liberiaan, kun heillä ei ole lääketieteellistä koulutusta huolehtia kuolevista afrikkalaisista?
Terveysjärjestö WHO alkoi lobbaamaan Ebola-virusta ISIS terroristien kautta

Maailman terveysjärjestö WHO alkoi lobbaamaan Ebola-virusta ja sen tulevia rokotuksia, yhdistämällä ISIS -terroristit Ebolaan. Daily Mail -lehti satuili otsikolla "Isis-taistelijat saattavat kantaa Ebolaa".
https://www.youtube.com/watch?v=aOMgenA56Ao
Yhdysvaltalainen propaganda uutiskanava CNN välitti vuoden 2014 lopussa vastaavanlaista propagandaa, että ISIS terroristit levittäisivät Ebolaa.
Kaiken tämän takana on suurien lääketehtaiden liikevaihdon turvaaminen, jolla he haluavat nyt saada tarpeeksi suuren pelon aikaiseksi tällä Ebola uutisoinnilla ja tämä CIA:n organisoima terrorijärjestö ISIS on katsottu nyt tehokaaksi tavaksi lobata tulevia Ebola-rokotteita eteenpäin.
Eräs suomalainen mikrobiologian lääkäri paljasti jo 2013 vuoden lopulla, kuinka suomen tartuntalakia valmisteleva ryhmä on pohtinut nyt SOTE 2015 uudistuksessa Suomen kansalaisten yksilön vapauden ja vastuun rajoja. Rivien välistä tämän voi nähdä käsittävän vaikka Ebola "pakkorokotuksia". Mutta jostain syystä mediassa ollaan Ebola -rokotuksista vielä toistaiseksi oltu hiljaa, vaikka Teksasin yliopistossa kehitettiin jo yksi rokote.

Kaiken tämän maailmanlaajuisen rokotehysterisoinnin takana piilee lääketeollisuuden (BIG PHARMA) voitontavoittelu. Vaikka tavoitteena on miljardivoitot, niin massarokotus suunnitelman salainen poliittinen agenda on valtaväestön massasterilointi, koska syntyvyys tulisi saada loppumaan. Vuonna 1992 säädetty YK:n agenda 21 = Globaali depopulaatio-ohjelma. Mutta samoin myös ilmastonmuokkauksella aikaansaatu ilmastonmuutos on todellisuudessa tätä samaa eugeniikkaa.

Jos sen jo nykyinen ympäristöministeri Sanni Grahn-Laasonen ilmoitti, että kuinka Suomesta on tavoite saada hiilineutraali valtio vuoteen 2050 mennessä ja Niin ei tässä mistään mustikkapensaiden pelastamisesta ole kyse. Ihminen kun sattuu hengittämään hiilidioksiidia ja jos Suomi on luvannut vähentävänsä hiilipäästöjään 90 % vuoteen 2050 mennessä, niin jo on merkillinen death-line. Se kun sattuu olemaan juuri sama vuosi death-line, kuin muutamilla pankkiirien ajatushautomoilla (Committee of 300). Tämän kolmensadan komitean ajatushautomon tavoite olisi saada maailman väestöä vähennettyä 90 % vuoteen 2050 mennessä.

Järjenkäyttö on sallittua ja suositeltavaa kyseenalaistaessa sen, että kiinnostaako tätä pankkiirien valtaeliittiä soiden ja mustikkapensaiden pelastaminen ? Massa-depopulaatio on heidän salainen tavoite ja sillä he ovat kehittäneet vielä suuren bisneksen, kun toiminta ajetaan "ilmastonmuutoksen" nimisen toimintaohjelman kautta.
On vielä häikäilemätöntä huomata, kuinka erityisesti Vihreät puolueena onottanut tämän "ilmastonmuutoksen" virallisiin vaaliteemoihinsa. Mutta moniko puolue olisi valmis ottamaan Suomen väestöön kohdistetun eugeniikan virallisiin puolueohjelmiin, kun tämä "rakenneuudistus" käytännössä jo on sitä.
Lähteet: http://www.iltalehti.fi/ulkomaat/2015010218970161_ul.shtml
http://www.nationalreview.com/article/389678/weird-science-cnn-calls-ebola-isis-bio-agents-tim-cavanaugh
http://www.vihreat.fi/node/8699
http://nwohavaintoja.blogspot.fi/2013/09/pakkorokotuslakia-juonitaan-parhaillaan.html
http://www.tesso.fi/kasvot/anni-virolainen-julkunen-mikrobit-askeleen-ihmista-edella
http://www.nationalreview.com/article/389678/weird-science-cnn-calls-ebola-isis-bio-agents-tim-cavanaugh
http://www.vihreat.fi/node/8699
http://nwohavaintoja.blogspot.fi/2013/09/pakkorokotuslakia-juonitaan-parhaillaan.html
http://www.tesso.fi/kasvot/anni-virolainen-julkunen-mikrobit-askeleen-ihmista-edella
Suomen mediassa on jatkunut Ebolalla pelottelu informaatiosodan muodossa, kun Helsingin uudenmaan sairanhoitopiiri HUS julisti iltapäivälehtiä myöten, kuinka Helsinki-Vantaan lentoasemalle saapui Liberiasta Ebola epäilty potilas. HUSLABin turvalaboratoriossa alkuillasta valmistuneen ebolatestin tulos oli negatiivinen.

Tapaus oli jo toinen laatuaan tämän kuun sisällä ja edelliselläkään kerralla tuolla Suomen Ebola epäilyllä ei ollut Ebolaa.
Nyt Suomen Terveyden ja Hyvinvoinninlaitos THL miettii jatkossa, että mitähän ulkoapäin käskettyä propagandaa tästä Ebolasta seuraavaksi uutisoitaisiin eteenpäin.

THL on saanut eräältä suurelta lääkeyhtiöltä Ebola-rokotteiden lobbausrahaa muutamia miljoonia. Nyt käynnissä oleva vaihe keskittyy Suomen tiedotusvälineissä pelotteluun ja tätä koko prosessia valvotaan myös vaikutusvaltaisempien viranomaisten taholta. Kuten sikainfluenssa -epidemia oli myös täysin valheilla tekaistu ja sen koko feikkiepidemian tarkoitus oli saada pakotettua koko Suomen väestö ottamaan sikainfluenssarokotus. Mutta tuo suunnitelma meni lopulta karille.

Mutta tällä kerralla Ebola-piikkejä ollaan jo tilaamassa Suomeen ja 2015 vuoden SOTE-uudistukseen ollaan kirjattu pykälä, jossa mahdollisen WHO:n julistaman pandemian johdosta koko Suomen kansa joutuisi pahimmillaan pakkorokotusten uhreiksi. Mikä tarkoittaisi koko Suomen väestön turvallisuuden vaarantumista, jos näillä immuniteettisuojan heikennysrokotteilla aiotaan tuhota koko Suomen kansan terveys ja vastustuskyky.

Ebolalla pelottelu ei vaan tule toimimaan, jos tietää mikä on tämän Ebolalla pelottelun poliittinen agenda. USA:n hallituksen suunnitelma on tappaa maailman väestöstä 90 % vuoteen 2050 mennessä.USA:n hallitus tekee sitä nyt myös kehittämällään ja patentoimalla nanobioaseella EBOLA.

Vaikka he ovat jo huhujen mukaan rakennuttaneet itselleen laajoja maanalaisia turvabunkkereita pitkin Yhdysvaltoja, niin depopulaatiohanke on saanut jatkuvasti takapakkia.
Vaikka he ovat jo huhujen mukaan rakennuttaneet itselleen laajoja maanalaisia turvabunkkereita pitkin Yhdysvaltoja, niin depopulaatiohanke on saanut jatkuvasti takapakkia.

Maailmanlaajuisessa väestönvähennyshankkeessa ensimmäisenä listalla ovat tietenkin eliitin mukaan köyhimmät maanosat kuten Afrikka. Juuri tästä syystä Afrikkaa tuodaan esille tämän Ebolan suhteen mediassa erityisen paljon. Tästä 2009-2010 patentoidusta Ebola-viruksesta on olemassa monia versioita, joista rankin virusmalli olisi tarkoitus pistää kulkeutumaan ihmisiin juuri Afrikassa.

Miten Zairen Ebola pääsi Länsi-Afrikkaan, noin 3 500 kilometrin päästä, jossa se oli ensimmäiseksi tunnistettu vuonna 1976?
Tämä on hyvä kysymys Washingtonille, mutta siihen emme saa mitään vastauksia? Eikä varmaan ihan heti, sillä kesti yli 62 vuotta paljastaa Guatemalan kuppakokeilut yleisölle, eikä sekään tapahtunut Yhdysvaltain hallituksen vaan lääketieteen historioitsijan toimesta.
"EBOLA-keksinnön yhteenveto"-osassa patenttidokumentti selvästi myös väittää, että USA:n hallituksella on "omistusoikeus" kaikkiin Ebola-viruksiin.
"...invention relates to the isolated EboBun virus that morphologically and phylogenetically relates to known members filoviridae... In another aspect, the invention provides an isolated hEbola EboBun virus comprising a nucleic acid molecule comprising a nucleotide sequence selected from the group consisting of: a) a nucleotide sequence set forth in SEQ ID NO: 1; b) a nucleotide sequence that hybridizes to the sequence set forth in SEQ ID NO: 1 under stringent conditions; and c) a nucleotide sequence that has at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to the SEQ ID NO:
1. In another aspect, the invention provides the complete genomic sequence of the hEbola virus EboBun."
Tutkijat väittävät, että tappavat taudit kuten Ebola ja AIDS ovat USA:n hallituksen kehittämiä nanobioaseita, joita on "kokeiltu" afrikkalaisiin. HIV-viruksen ja Ebola-viruksen levittämisellä pyritään tappamaan aluksi afrikkalaiset.
Tässä dokumenttielokuvassa paljastetaan miten pieni itsekäs ahne psykopaattijoukko on ohjannut maailman menoa nykyhistorian aikana ja miten se on yhä tänäpäivänä poliittisen vallan takana, miten se ohjaa maailmaa ja mikä on sen paha suunnitelma ihmiskunnalle.
Denverin lentokentällä on esitetty piirroksin suunnitelma ihmiskunnan järjestelmällisestä tappamisesta.

.jpg)
Kuva Denverin lentokentältä, jossa kuva kertoo suunnitelmasta. USA:n hallitus on pyrkinyt tappamaan näillä viruksillaan ensin kaikki afrikkalaiset.

Tetanus vaccines found spiked with sterilization chemical to carry out race-based genocide against Africans
Learn more: http://www.naturalnews.com/047571_vaccines_sterilization_genocide.html#ixzz3IgRSDk5W
(NaturalNews) Tetanus vaccines given to millions of young women in Kenya have been confirmed by laboratories to contain a sterilization chemical that causes miscarriages, reports the Kenya Catholic Doctors Association, a pro-vaccine organization.
https://www.lifesitenews.com/news/a-mass-sterilization-exercise-kenyan-doctors-find-anti-fertility-agent-in-u
http://www.naturalnews.com/041345_cdc_polio_vaccine_sv40.html
http://www.naturalnews.com/047358_ebola_outbreak_human_meat_immune_response.html
http://www.naturalnews.com/029911_vaccines_bill_gates.html

http://www.naturalnews.com/046347_ebola_vaccine_genetically_engineered_virus_depopulation.html
https://www.youtube.com/watch?v=oi1hXM3DYG8
A whopping 2.3 million young girls and women are in the process of being given the vaccine, pushed by UNICEF and the World Health Organization.
"We sent six samples from around Kenya to laboratories in South Africa. They tested positive for the HCG antigen," Dr. Muhame Ngare of the Mercy Medical Centre in Nairobi told LifeSiteNews. "They were all laced with HCG."
Chemical causes a woman's body to destroy its own fetus with vaccine-induced antibodies
HCG is a chemical developed by the World Health Organization for sterilization purposes. When injected into the body of a young woman, it causes a pregnancy to be destroyed by the body's own antibody response to the HCG, resulting in a spontaneous abortion. Its effectiveness lasts for years, causing abortions in women up to three years after the injections.Dr. Ngare explained "...this WHO campaign is not about eradicating neonatal tetanus but a well-coordinated forceful population control mass sterilization exercise using a proven fertility regulating vaccine."
The Kenyan government, of course, insists the vaccine is perfectly safe. Dr. Tabu of Kenya's Health Ministry even told the media that because some young women are still having babies, the vaccine therefore must not contain any sterilization agent. However, this claim belies the fact that HCG doesn't work 100% of the time. It only sterilizes the majority of those injected with it, not all of them.

More importantly, the Kenyan Catholic Church is a pro-vaccine organization. "What reason do the Catholic doctors have for lying?" asked Dr. Ngare as reported in the LifeSiteNews article linked above. "The Catholic Church has been here in Kenya providing health care and vaccinating for 100 years for longer than Kenya has existed as a country."
In other words, the very group exposing the sterilization agenda of the tetanus vaccines is in fact a pro-vaccination group. Yet even they have now come to realize the horrifying truth: vaccines are the perfect vector for governments to deviously insert covert chemical or viral agents which are never revealed to the public.
The smoking gun: a five-shot course over two years
What really raised red flags about this so-called tetanus vaccine was the highly unusual inoculation schedule. This vaccine demanded five shots over two years -- a schedule that isn't used for tetanus."The only time tetanus vaccine has been given in five doses is when it is used as a carrier in fertility regulating vaccines laced with the pregnancy hormone, Human Chorionic Gonadotropin (HCG) developed by WHO in 1992." explained Dr. Ngare.
Furthermore, the vaccine was only being given to women of child-bearing years, not men or women beyond the age of fertility.
As Dr. Ngare explains, the same vaccine sterilization campaign was used in 1993 in Mexico and both Nicaragua and the Philippines in 1994. WHO attempted to bring it to Kenya in the 1990's, Ngare says, but the effort was stopped by the Catholic Church.
According to Brian Clowes of Human Life International, the United Nations is not refuting the laboratory testing and confirmation of HCG in the vaccines. Instead, it claims some vaccines were "contaminated" in the manufacturing process -- an absurd claim that no reasonable person would believe because HCG should never even be anywhere near a vaccine manufacturing operation unless someone put it there deliberately.
LifeSiteNews reports that it:
has obtained a UN report on an August 1992 meeting at its world headquarters in Geneva of 10 scientists from "Australia, Europe, India and the U.S.A" and 10 "women's health advocates" from around the world, to discuss the use of "fertility regulating vaccines." It describes the "anti-Human Chorionic Gonadotropin vaccine" as the most advanced.
Read the full report from LifeSiteNews at:
http://www.lifesitenews.com/news/a-mass-ster...
https://www.youtube.com/watch?v=ogM3S1jimIY
United Nations, WHO and UNICEF all engaged in vaccination genocide
You will not see this news reported by any mainstream media outlet in the United States. All truth about vaccines is censored, even if the truth is that the United Nations is deliberately engaged in a campaign of vaccine genocide against people of Africa.What is happening in Kenya is a crime against humanity, and it is a crime committed with deliberate racial discrimination. Normally, the liberal media in the United States would be all over a story involving racial discrimination and genocide -- or even a single police shooting of a black teenager -- but because this genocide is being committed with vaccines, the entire mainstream media excuses it. Apparently, medical crimes against black people are perfectly acceptable to the liberal media as long as vaccines are used as the weapon.
As this story clearly demonstrates, "vaccine violence" is very real in our world. Vaccines are the perfect weapon for population control for several reasons:
1) Nobody really knows what's in them.
2) They can be easily spiked with hidden chemicals.
3) They can be administered under the cover of "public health."
4) All governments and establishment media will deliberately collaborate with the genocide in order to protect vaccines from being recognized as medical weapons against women.
Thus, vaccines can be routinely used to inject populations with birth control chemicals or even stealth cancer viruses. In fact, this is exactly what happened to as many as 98 million Americans during the mass polio vaccinations of the 1960's and 70's. The CDC even documented the "accidental" injection of millions of Americans with the cancer-causing SV40 simian virus, but the agency scrubbed all that history from its website in 2013.
In Kenya today, government authorities also claim the sterilization chemical was an "accidental" contamination. That's the excuse that can always be used as a cover story in weaponized vaccination schemes, where governments deliberately taint vaccines with known chemicals that end life, promote cancer or cause spontaneous abortions.
Vaccines as weapons = Medical crimes against humanity
The deliberate adding of HCG to vaccines without full disclosure to the population is a heinous violation of human rights and human dignity. Here are just a few of the crimes now being committed against humanity under the guise of vaccinations:CRIME #1) No informed consent. None of these women in Kenya were told the truth that they were being injected with a sterilization chemical designed to cause infertility.
CRIME #2) Race-based genocide. The targeting of Kenyan women with this vaccine is a deliberate selection based on their race. By any reasonable standard, this would be called a racially-motivated hate crime resulting in genocide.
CRIME #3) The deliberate killing of a human being. The spontaneous abortions caused by these HCG-spiked vaccines results in the ending of a human life inside the mother's body. These killings take place without the consent or permission of the mother, nor any opportunity for defense of the life of the unborn child.
CRIME #4) Violation of Geneva Convention limitations on medical experimentation. All these Kenyan women injected with this vaccine are being used as human guinea pigs in a covert, criminal medical experiment. None of these women voluntarily signed up for this medical experiment, nor were they even informed. This is a medical crime against human beings.
CRIME #5) Crimes against women. Only women were selected for this targeted sterilization vaccine effort, proving that this is not only a race-based crime but also a gender-based crime against women.
If you add all this up, you've got weaponized vaccines being intentionally spiked with a known sterilization chemical developed by the WHO, then deployed in a racially-motivated genocidal manner that targets women to be used in an illegal medical experiment administered via vaccine inoculations.
When administered via vaccines, genocide and murder are apparently not news
Yet, despite all this, the mainstream media is perfectly okay with this activity. The World Health Organizations endorses it. The United Nations organizes it. Governments help fund it. Vaccine-pushing scientists excuse it. Media outlets cover it up and censor the story, hoping you don't read Natural News or Life Site News to learn the truth.When pharmacies in your neighborhood push flu shots and other vaccines, they don't tell you they are part of a branch of medicine steeped in genocide, racially-motivated hate crimes and a medical war on women. They don't tell you that flu shots still contain toxic mercury at concentrations 100 times the mercury found in ocean fish. They don't tell you anything about what's in those vaccines for the same reason that women in Kenya are never told what's in them, either.

Vaccines are the perfect weapons against women and children
The truth is that vaccines are easily deployed as weapons against humanity under the false cover story that they are saving humanity. What better way to pursue deliberate chemically-induced population control than to convince people they are being injected "for their own good?"This is precisely why Bill Gates famously said:
The world today has 6.8 billion people... that's headed up to about 9 billion. Now if we do a really great job on new vaccines, health care, reproductive health services, we could lower that by perhaps 10 or 15 percent.
Why would Bill Gates be talking about vaccines REDUCING human population if vaccines didn't secretly contain sterilization agents? Remember, Gates is the same person who has funded all sorts of sterilization technologies including one that blasts men's scrotums with high-intensity sound waves to make them infertile.

Top tools for human depopulation
Gates is part of a covert medical cabal that believes aggressive human depopulation is urgently necessary to save the planet. This group, which includes many scientists and virologists, believe that the most effective tools for human depopulation are:1) Vaccines which are covertly spiked with sterilization chemicals.
2) Genetically engineered viruses with a high mortality rate, possibly engineered to target specific races and genetic profiles.
For example, Dr. Charles Arntzen, head of The Biodesign Institute for Infectious Diseases and Vaccinology recently joked about using an engineered virus to cull the human population, saying "That's the answer! Go out and use genetic engineering to create a better virus. (laughter) Twenty-five percent of the population is supposed to go in Contagion."
As I wrote on October 22, 2014, many virologists believe humans are nothing more than a "parasite" to be consumed by viruses which are the planet's "immune response" to human overpopulation. Here's a passage from the book "The Hot Zone" by Richard Preston, summarizing the way these scientists think:
...the earth is mounting an immune response against the human species. It is beginning to react to the human parasite, the flooding infection of people, the dead spots of concrete all over the planet, the cancerous rot-outs in Europe, Japan and the United States, thick with replicating primates [i.e. humans], the colonies enlarging and spreading and threatening to shock the biosphere with mass extinctions.
Perhaps the biosphere does not "like" the idea of five billion humans. Or it could also be said that the extreme amplification of the human race, which has occurred only in the past hundred years or so, has suddenly produced a very large quantity of meat, which is sitting everywhere in the biosphere and may not be able to defend itself against a life form that might want to consume it...
The earth's immune system, so to speak, has recognized the presence of the human species and is starting to kick in. The earth is attempting to rid itself of an infection by the human parasite.
What's extraordinary in all this -- both with vaccines and viruses engineered as weapons -- is how the most influential people in the scientific community have come to view humanity as an enemy to be destroyed via tools of medicine and science. Frighteningly, modern medical science has the tools to carry out its genocidal assaults on humankind through "accidental" releases of deadly viruses or "accidental" contamination of vaccines with sterilization chemicals.
The evidence of deliberate sterilization chemicals in United Nations vaccines raises the obvious question: Was the recent Ebola outbreak in West Africa also intentional? And what else might scientists, vaccine pushers, world health authorities and governments have in mind for human depopulation in the years ahead?
Is there already something in the food supply that causes sterilization? The answer is a definite YES, and just like the pandemic viruses, it too is genetically engineered.

The five vectors for destroying humanity
These are the vectors for the science-based genocidal assault on humanity:1) Vaccines
2) Viruses
3) Food
4) Water
5) Chemtrails (i.e. atmospheric deployment of chemicals)

https://www.youtube.com/watch?v=L5is16A8pfw

https://www.youtube.com/watch?v=v-zRREd8DZQ
https://www.youtube.com/watch?v=baQlv1th7y4

Kultananopartikkelit voivat ohjata aivojen käyttäytymistä

Nyt se on sitten ihan MIT (Massachusetts Institute of Technology) myöntänyt kuinka kultananopartikkeleilla voidaan ohjata aivojen käyttäytymistä.

Tätä
keksintöä julistetaan nyt mm. Guardianissa uusimmaksi tieteelliseksi
menetelmäksi parantaa mm. kasvaimia. Chicagon yliopiston tutkijat ovat
huomanneet, kuinka sillä voidaan ohjata aivojen hermosoluja.
Toinen tutkijoiden ryhmä käytti kokeissaan nano kokoista rautaoksidia, jotka kuumenevat kun magneettikenttä osuu siihen.

Suihkukoneista on jo vuosikaudet pudotettu näitä samoja myrkyllisiä nanopartikkeleita sisältäviä kemikaaleja alas ja edelleenkään asiasta ei saa julkisesti keskustella. Koska niiden takana on mm. sotilasliitto Nato ja toimintaa organisoi rikolliset pankit.
Nyt yhdysvaltalaisen yliopiston tutkijat myöntäjät, kuinka nanopartikkeleilla voidaan ohjata aivojen toimintaa. Mutta kemikaalivanoista ei edelleenkään saa puhua julkisesti, koska siellä vaikuttaa niin suuri rikollinen pääoma. Itseasiassa se asia mitä kemikaalivanoissa salataan eniten on niiden käyttötarkoitus (depopulaatio). Sillä tämä ilmateitse harjoitettava eugeniikka on täysin rikollista toimintaa, jonka organisoijat tulisi laittaa vastuuseen.
Kemikaalivanat ovat edelleen kuuma ja kiistelty puheenaihe. Kerran yksi kemikaalivana suihkukoneen pilotti unohti myrkytyshanat auki ja se tallentui videolle.
Toinen tutkijoiden ryhmä käytti kokeissaan nano kokoista rautaoksidia, jotka kuumenevat kun magneettikenttä osuu siihen.
Suihkukoneista on jo vuosikaudet pudotettu näitä samoja myrkyllisiä nanopartikkeleita sisältäviä kemikaaleja alas ja edelleenkään asiasta ei saa julkisesti keskustella. Koska niiden takana on mm. sotilasliitto Nato ja toimintaa organisoi rikolliset pankit.
Nyt yhdysvaltalaisen yliopiston tutkijat myöntäjät, kuinka nanopartikkeleilla voidaan ohjata aivojen toimintaa. Mutta kemikaalivanoista ei edelleenkään saa puhua julkisesti, koska siellä vaikuttaa niin suuri rikollinen pääoma. Itseasiassa se asia mitä kemikaalivanoissa salataan eniten on niiden käyttötarkoitus (depopulaatio). Sillä tämä ilmateitse harjoitettava eugeniikka on täysin rikollista toimintaa, jonka organisoijat tulisi laittaa vastuuseen.
Kemikaalivanat ovat edelleen kuuma ja kiistelty puheenaihe. Kerran yksi kemikaalivana suihkukoneen pilotti unohti myrkytyshanat auki ja se tallentui videolle.
Mutta vain hyvin harvat lentäjät uskaltavat puhua aiheesta, koska heitä on jopa uhkailtu sen johdosta jos uskaltavat ottaa asian puheeksi.

Läpäisyelektronimikroskopian avulla otettuja kuvia (a, b, ja c) piidioksidien nanopartikkeleista, joiden keskimääräinen halkaisija on kuvissa (a) 20 nm, (b) 45 nm, ja (c) 80 nm. Pyyhkäisyelektronimikroskoopin avulla otettu kuva (d) samasta näytteestä joka on kuvassa (b).
Nano partikkelit
Tavallisille kaduntallaajille ei paljon kerrota näistä "nanopartikkeleista" että mitä ne on ja mihin niillä pyritään.
Yleisimmin käytetty nanopartikkeli on titaanioksidi. Koe-eläin hiirille tehtiin testi joissa heille juotettiin titaanioksidia sisältävää vettä. Niin huomattiin että viidessä vuorokaudessa heidän DNA:n kromosomit vaurioituivat. Tässä jo yksi esimerkki nanopartikkeleiden vaarallisuudesta.
GlaxoSmithKlinen valmistamissa sikainfluenssarokotuksissa oli skvaleeniin tehty nanopartikkeliseos. Eli näin ollen myrkyt eivät poistu lainkaan elimistöstä, koska ne voivat mennä suoraan aivoihin ja näin estää mm. lapsen aivojen kehitystä.
Turussa toimiva biotekniikan keskus tutkimusryhmän käyttämiä nanopartikkeleita käytetään mm. löytämään syöpäsolu, jossa hiukkanen ohjelmoidaan löytämään se.

Tätä nanoteknologiaa ei kuitenkaan eräästä syystä käytetä BIG PHARMA:n syöpätutkimuksissa syövän parantamiseen. Vaan siinä keskitytään BIG PHARMA:n etujen, eli liiketoiminnan turvaamiseen. Mikä tarkoittaa luonnollisesti sitä, että mitä sairaampia ovat potilaat, niin sitä enemmän lääketeollisuuden mafian kassakoneet kilisevät ja syöpä on näistä liiketoimintamalleista miltei tuottoisin. Syövän kun voi parantaa mm. ruokasoodalla mutta tätä ei BIG PHARMA halua kenellekään kertoa.

Yllä on yksi havainne kuva kemikaalivanojen aiheuttamien alumiinimyrkkyjen vaikutuksesta aivoihin. Ylempänä on normaalit aiot ja alempana kemikaalivanoista taivaalta tiputetuille alumiinimyrkyille altistuneet aivot.

Alumiinin sisältämät myrkyt altistavat mm. alzheimerille. Vuonna 2008 tehtiin tutkimus jossa huomattiin että erityisesti alueilla joissa juomavedessä on runsaasti alumiinia, esiintyy poikkeuksellisen paljon alzheimerin tautia.
All five of these vectors present "opportunities" for genocidal scientists to achieve their goal of human sterilization and depopulation. That is precisely why anyone who wishes to survive the great human culling now under way must take extraordinary steps to isolate themselves from institutionally-produced food, water and medicine. The only safe food, water and medicine is that which was produced independently and far outside the control of Big Food, Big Ag and Big Pharma.
Don't drink the city water without filtering it first, and read my laboratory testing results for all popular water filters at www.WaterFilterLabs.com
Don't eat factory-produced food. Don't allow yourself to be injected with weaponized vaccines. Don't take Big Pharma's deadly medicines. Be smart by being skeptical about the claimed "safety" of all those things created by institutions and authorities that quite literally want to kill off a significant percentage of the existing world population.
If you're smart and resourceful, you might just survive this great human culling. On the other hand, those who anxiously line up to be injected with the seasonal flu shots are all admitting they are too stupid and gullible to last long in a world where "science" has declared a covert war on human life.
Learn more: http://www.naturalnews.com/047571_vaccines_sterilization_genocide.html#ixzz3IgRghbqA
https://www.youtube.com/watch?v=ogM3S1jimIY
Kuva Denverin lentokentältä, jossa kuva kertoo suunnitelmasta. USA:n hallitus on pyrkinyt tappamaan näillä viruksillaan myös alkuperäiskansat.

USA:n hallitus haluaa tappaa ensin afrikkalaiset ja alkuperäiskansat, mutta se on käynyt tappamaan myös muitakin.
USA:n hallitus on kehittänyt hiv-viruksen ja ebola-viruksen laboratoriossa.
USA:n hallitus on patentoinut sekä hiv-viruksen ja ebola-viruksen, joilla USA on käynyt sotaan ihmiskuntaa vastaan.

Kuva Denverin lentokentältä, jossa kuva kertoo suunnitelmasta. USA:n hallitus haluaa tappaa näillä viruksillaan myös pohjoismaalaiset ja kaikki valkoiset rodut.
https://www.youtube.com/watch?v=ogM3S1jimIY
Scientists allege deadly diseases such as Ebola and AIDS are bio weapons being tested on Africans. Other reports have linked the Ebola virus outbreak to an attempt to reduce Africa’s population. Liberia happens to be the continents’s fastest growing population.

Miksi Yhdysvaltain hallitus on kehittänyt Ebola-viruksen laboratorioissaan?
Miksi Yhdysvaltain hallitus on patentoinut tämän Ebola-keksintönsä?
Miksi Yhdysvaltain hallitus on vaatinut monopolin "keksintöönsä"?
Miksi USA omistaa tämänkin ihmiskunnan tappamiseen kehittämänsä patentin, "jolla" tapetaan ihmiskuntaa parhaillaan?
http://www.youtube.com/watch?v=Vtk_3S33Ag0
Maailman terveysjärjestön eli WHO:n arvion mukaan maailmassa on monta kymmentä miljoonaa HIV-tartunnan saanutta. Tilanne on pahin Afrikassa Saharan eteläpuolella, jossa tartunnan saaneita lasketaan olevan useita miljoonia.
Suomessa lääkärit ovat ilmoittaneet marraskuun 1993 loppuun mennessä 562 HIV-tartuntaa. Näistä neljäsosalla tartunta on kehittynyt AIDS-vaiheen taudiksi. Suomessa on kuollut AIDS:iin useita satoja.
Siirtykäämme ajassa yli viisikymmentä vuotta taaksepäin. WHO kirjoitti tiedotuslehdessään "Bulletin of the World Health Organisation" numerossa 47 sivulla 259 vuonna 1972:
"Tulisi tehdä yritys sen toteamiseksi, voivatko virukset itse asiassa panna liikkelle valikoivia vaikutuksia vastustuskyvyn toimintaan. Tulisi katsoa sitä mahdollisuutta, että voitaisiin heikentää vastustuskyvyn reaktiota virukselle, jos tarttuva virus vahingoittaa enemmän tai vähemmän valikoivasti virukselle reagoivan solun."
Selvällä suomenkielellä sanottuna WHO kehoitti kehittämään juuri AIDS:in kaltaisen viruksen!
WHO kertoi lisäksi, että jos uusi viruksen luomus toimisi, niin monista kauhistuttavista ja kuolettavista tarttuvista viruksista voitaisiin tehdä jopa vielä kauhistuttavampia ja pahanlaatuisempia.
Eikös olekin omituista tekstiä terveysjärjestön kirjoittamaksi?
Ensimmäiset AIDS-tapaukset ilmaantuivat 1970-luvulla ja pian siitä tuli maailmanlaatuinen ongelma. Asiantuntijat kertoivat tiedotusvälineissä, että HIV-tartunta on peräisin vihreästä apinasta Afrikassa.
AIDS alkoi kuitenkin samanaikaisesti Yhdysvalloissa, Haitissa, Brasiliassa ja Keski-Afrikassa, joten tuon vihreän apinan on täytynyt olla suihkukonelentäjä!
Vihreän apinan solujen geenirakenteen tutkimus onkin osoittanut, ettei ole geneettisesti mahdollista siirtää AIDS-virusta luonnollisella tavalla apinoista ihmiseen.
Vuonna 1987 tapahtui suuri paljastus. Arvostettu sanomalehti "London Times" kirjoitti 11.5. etusivulla:
"Isorokkorokote pani alulle AIDS-viruksen."
HIV-tartunnat vastasivat täysin WHO:n rokotusohjelmaa Haitissa, Brasiliassa ja Afrikassa, jossa Zairessa eniten rokotuksia saaneena oli myös eniten HIV-tartuntoja.
Artikkeli sai alkunsa siitä, kun WHO itse alkoi epäillä, oliko sen rokotusohjelmalla yhteyttä AIDS-epidemiaan.
Se vuokrasi ulkopuolisen tutkijan ottamaan asiasta selvää. Tutkija löysi selvän yhteyden HIV-tartuntojen ja rokotusohjelman välillä. WHO kuitenkin salasi tutkimusraportin, jolloin tuo tutkija kääntyi "London Times" -lehden puoleen. Paitsi Lontoossa, sokkiuutinen
joutui täydelliseen uutispimentoon, eikä sitä julkaistu päätiedotusvälineissä missään muuallapäin maailmaa.
Vaikka "London Times" -lehden artikkeli sanoikin, että
isorokkorokotusohjelma laukaisi Afrikassa piilevän taipumuksen saada AIDS, on myöhemmin todettu, ettei ole minkäänlaista tieteellistä todistetta siitä, että AIDS-virus olisi ollut Afrikassa ennen WHO:n rokotusohjelmaa.
Totuuden tuojina AIDS:sta ovat olleet etummaisina amerikkalaiset lääkärit William Douglass ja Theodore Strecker.
Tri Streckerin perinpohjaiset tutkimukset osoittivat, että Kansallinen
syöpäinstituutti ja WHO loivat yhteistyössä AIDS-viruksen laboratorioissaan Fort Detrickissä. AIDS-virus syntyi yhdistämällä kaksi tappavaa retrovirusta eli lehmissä esiintyvän leukemiaviruksen (BLV) ja lampaissa esiintyvän visnaviruksen toisiinsa ja kasvattamalla niitä sitten ihmisen kudosviljelmässä. Tri Douglass väittää lisäksi, että HIV-virus ei ole huonosti tarttuva virus. Se voi elää jopa 10 päivää kuivalla lautasella.
Miksi AIDS on sitten pesiytynyt homoseksuaaleihin? Vanhoista
verinäytetutkimuksista on havaittu, että Yhdysvalloissa ei ollut
olemassa AIDS-virusta ennen vuotta 1978.
AIDS:in äkillinen leviäminen on osoitettu olevan selvästi yhteydessä homoseksuaaleille annetun hepatiitti B-rokotusohjelman kanssa. Jostain selittämättömästä syystä Puolassa syntynyt Venäjällä opiskellut tri W. Szmuness pääsi New Yorkin
veripankin päälliköksi muutettuaan maahan vasta vuonna 1969.
Hän määräsi säännöt hepatiittirokotetutkimukselle, joissa hyväksyttiin 20-40 vuotiaat mieshenkilöt, jotka EIVÄT olleet yksiavioisia!
Voitko keksiä tällaiselle vaatimukselle mitään muuta syytä, kuin taudin levittäminen?
Tri Szmuness kuoli myöhemmin vieden salaisuuden mukanaan.
Näin AIDS levisi myös Yhdysvaltoihin. Roy Livesey kirjassaan
"Understanding the New World Order" (Uuden maailmanjärjestyksen ymmärtäminen) kertoo raportissa vuodelta 1980, jossa Rooman klubin jäsen ja biologi sanoo eräässä amerikkalaisessa yliopistossa:
"On tärkeää, että Yhdysvallat vähentää väkilukuaan kahdella kolmasosalla seuraavan 50 vuoden aikana."
Väkiluvun vähennys on siellä tosiaan juuri käynnissä.
Tri Theodore Strecker lähetti kirjeitä muutamiin maailman
arvostetuimmista lääketieteellisistä lehdistä, mutta yksikään ei
julkaissut hänen kirjoitustaan. Esim. Lancet -lehti vastasi hänelle,
että sen kirjeosasto on liian täynnä sinne saapuneita kirjoituksia.
Viime aikoine on kuitenkin ollut maailman lehdissä yksittäisiä
kirjoituksia, joissa asiantuntijat ovat sanoneet, että AIDS on luotu
laboratoriossa.
AIDS:in alkuperästä on saatavana hyvin mielenkiintoinen video "The Strecker Memorandum", jota voi tilata osoitteesta: The Strecker Group, 1501 Colorado Blvd, Eagle Rock, CA 90041, USA. Aihetta valaisee hyvin myös kirja Dr Alan Cantwell: "AIDS And The Doctors of Death", Aries Rising Press, Los Angeles, USA.
WHO ei varmaankaan myönnä osallisuuttaan AIDS:in levittämisessä. Jo korvausvaatimukset olisivat liian suuret puhumattakaan maineen menetyksestä. Vaikka maailman väkiluku onkin laskenut AIDS:in johdosta, ei maailmaa yhdentävien taustavoimien voida virallisesti osoittaa olevan AIDS-ongelman takana.
On kuitenkin helppo todeta, että AIDS:in syntymisessä ja leviämisessä on todella jotain mätää. Sitä todistavat myös perusteettomien apinateorioiden levittäminen virallisena totuutena maailman päätiedotusvälineissä, jotka ovat valtaeliitin
kontrollissa. Samanaikaisesti yhteys WHO:n rokotusohjelmaan salataan.
http://www.youtube.com/watch?v=fevYSHQoeD4
The Government Of The United States Of America As Represented By The Secretary, Department Of Health & Human Services, Center For Disease Control.
"The invention provides the isolated human Ebola (hEbola) viruses denoted as Bundibugyo (EboBun) deposited with the Centers for Disease Control and Prevention ("CDC"; Atlanta, Georgia, United States of America) on November 26, 2007 and accorded an accession number 200706291."
"The present invention is based upon the isolation and identification of a new human Ebola virus species, EboBun. EboBun was isolated from the patients suffering from hemorrhagic fever in a recent outbreak in Uganda."
https://www.lifesitenews.com/news/fbi-interested-in-texas-doomsday-ecologist-who-said-ebola-the-solution-to-h
https://www.youtube.com/watch?v=ogM3S1jimIY
Evidence Mounts: Ebola Made In The USA By Big Pharma & Dept Of Defense?
Sources claim the virus was given to civilians through UN vaccine programmes with sinister aims
The scientist who recently pointed to evidence that Ebola was created by pharmaceutical corporations at the highest echelons of power was backed up by another expert- who says the outbreak coincides exactly with UN vaccine campaigns in the region.
Dr. Cyril Broderick is a former professor of plant pathology at the University of Liberia’s College of Agriculture and Forestry, and is on tenure as an associate at Delaware University. Late last month he wrote an op-ed in the Liberian Daily Observer claiming that the Ebola virus currently raging in western Africa is a GMO, made in a lab by western pharmaceutical companies and administered to unsuspecting civilians through U.N vaccination programs. The Washington Post called it a wild conspiracy theory, but is there any basis to these shocking revelations?
The Claims:
1) Ebola is a man-made GMO, created for depopulation of the region, profit for big pharma, and to gain access to the region´s rich mineral wealth (under the guise of military peace-keeping).
2) Testing has been taking place on unsuspecting Africans through various respectable charitable organizations who pretend to be vaccinating against meningitis, polio, cholera, etc
3) Countries involved in this conspiracy against African civilians are the US, Canada, France,and the UK.
4) Organizations implicated are the World Health Organization (WHO), Medecins sin frontiers, UNICEF, US Center for Disease Control (CDC), National Institute of Health (NIH), and The US Army Medical Research Institute of Infectious Diseases (USAMRIID). Links to the Lóreal group, owned by The Rothschilds, and the Bill & Melinda Gates Foundation are also mentioned in depth.
The Accused
In his controversial article, Dr. Broderick made reference to book called Emerging Viruses: AIDS and Ebola – Nature, Accident or Intentional? (Leonard Horowitz, 1998), stating that the author “confirmed the existence of an American Military-Medical-Industry that conducts biological weapons tests under the guise of administering vaccinations to control diseases and improve the health of black Africans overseas.”
In the book mentioned by Dr. Broderick, Dr. Horowitz had indeed unearthed documents about a shocking study in which chimpanzees were affected with numerous viruses, used to produce hepatitis B vaccine doses. He asserts that these were administered to central African civilians along with gay men in New York City at precisely the time the HIV epidemic ´punctuated event´occurred. A full-length 3 hour video on Dr. Horowitz´s findings can be seen here.
Dr. Broderick also points out that Dr. Horowitz interviewed a Dr. Robert Strecker in the 1970s, and that during this meeting, “mention was made of Fort Detrick,´the Ebola Building´” (Frederick, Maryland) – over thirty years ago. Dr. Broderick´s damning accusations may sound like the ramblings of a lunatic, but unlike most wacky ´conspiracy theories´ on the net, many seem to be rooted in hard fact.
His claims are backed up by Yoichi Shimatsu, a Thailand-based science writer who organized public health seminars by leading microbiologists and herbalists during the SARS outbreak in Hong Kong and the avian influenza crisis across Southeast Asia. Shimatsu has also written an article which appeared in the Liberian Daily Observer (the country´s biggest newspaper)- pointing out that the Ebola outbreak coincided exactly with U.N vaccination programs in West Africa, and naming some very high-profile players.
The-Ebola-virus
Shimatsu states: “The earliest breakout in Guinea coincided with three major vaccine campaigns conducted by the World Health Organization (WHO) and the UN children’s agency UNICEF. At least two of the vaccination programs were implemented by Medicins Sans Frontieres (MSF, or Doctors Without Borders), while some of those vaccines were produced by Sanofi Pasteur, a French pharmaceutical whose major shareholder is the Rothschild Group.”
The plot thickens. Here is what Shimatsu has dug up from his investigations:
• The MSF-WHO project administered the anti-cholera vaccine Shanchol. The drug producer (Shanta Biotechnics in Hyderabad, India) is a wholly owned subsidiary of Sanofi Pasteur based in Lyon, France. Formerly known as Sanofi Aventis, the pharmaceutical is controlled by major shareholders L’Oreal and the Rothschild Group.
• The oral polio vaccine drive funded by UNICEF was based on a pathogen seed strain developed by this Rothschild-owned corporation, which operates the world’s largest polio vaccine production facility.
•The meningitis vaccine MenAfrVac was produced by the Serum Institute of India, owned by tycoon Cyrus Poonawalla, under development funding from the Bill and Melinda Gates Foundation. In 2013, a UNICEF drive in Chad with the same drug resulted in 40 child deaths from vaccine-linked symptom. MSF participated in the West African anti-meningitis project.
Dr. Broderick adds some extra names to these institutions and organizations indicted by Shimatsu: The UK’s GlaxoSmithKline, a Canadian pharmaceutical company called Tekmira, the US Center for Disease Control (CDC), The US Army Medical Research Institute of Infectious Diseases (USAMRIID), a well-known center for bio-war research, located at Fort Detrick, Maryland, and Tulane University, in New Orleans, USA, “winner of research grants, including a grant of more than $7 million the National Institute of Health (NIH) to fund research with the Lassa viral hemorrhagic fever.”
How and Why?
According to Shimatsu: “The reason for suspecting a vaccine campaign rather than an individual carrier is due to the fact that the Ebola contagion did not start at a single geographic center and then spread outward along the roads. Instead, simultaneous outbreaks of multiple cases occurred in widely separated parts of rural Guinea, indicating a highly organized effort to infect residents in different locations in the same time-frame…The simultaneous eruptions of this filovirus virus in widely separated zones strongly suggests that the virulent Zaire ebola strain (ZEBOV) was deliberately introduced to test an antidote in secret trials on unsuspecting humans.”
Shimatsu believes that the escape of Ebola from Guinea into neighboring Sierra Leone and Liberia indicates something went terribly wrong during the illegal clinical trials by a major pharmaceutical company. “Through the lens darkly, the release of Ebola may well have been an act of biowarfare in the post-colonial struggle to control mineral-rich West Africa,” he writes.
“The Guinea outbreak was not reported by WHO until 6 weeks after the initial round of infections in February, which is quite odd considering the armies of medical workers afield in the countryside during those three vaccine campaigns,” Shimatsu states. “Despite assurances from WHO and CDC that ebola is not transmitted through water or air, more than 100 nurses and doctors, including Sierra Leone’s top ebola expert, have died so far. Misinformation about ebola transmission is inexcusable when the 1995 Zaire outbreak was first spread by the washing of corpses.”
Shimatsu doesn´t claim that healthcare workers within these organizations are aware of this dark plot- of course they are not- but he does make some very shady connections:
•In 2008, the Rothschild-connected Sanofi Pasteur corp was accused of conducting secret trials of an untested H5N1 vaccine on 350 homeless people in Poland, killing at least 21 and causing the hospitalization of 200 others.
•This is the world’s third-largest pharmaceutical, under CEO Serge Weinberg. Weinberg scored a coup in wooing his new chief scientist Gary Nabel from his position as head of viral immunology research at the National Institutes of Health (NIH).
•Sanofi is a partner company of Sutro Biopharma based in San Francisco. Sutro´s CEO is John Freund, a former Morgan Stanley executive.
•There are two potential cures for Ebola issued by biotech companies ZMapp and Tekmira. “Whichever company gains approval from an FDA, ready to overlook the possibility of driving mutations, will be sure to win huge supplier contracts from the WHO and the US Department of Defense”, Shimatsu writes. “The dark horse in the foot race to profit from the Ebola panic is Sanofi Pasteur. .”
But, says Shimatsu, it´s not all about profit- it´s also about stealing mineral wealth and gaining control of a country gripped by fear and desperation. “After rural West Africans realized that vaccination programs coincided with the outbreak of Zaire Ebola, foreign-funded medical staffers were assaulted by angry mobs and an Ebola treatment center in Sierra Leone was burned to the ground. When medicine is exposed to be the problem and not a solution, the military has to be called in to quell public rebellion,” he explains. “Ebola detonated fear and loathing, and perhaps that is exactly the intended objective of a destabilization strategy.”
Not only that, but public anxiety fuelled by hysterical fear-mongering in the media is providing a very convenient excuse for right-wing governments (especially those in Western Europe) to crack down on immigration, stop flights and increase airport security in the name of the Ebola pandemic. Despite terrifying headlines, only a handful of westerners have contracted the disease – as a side note, isn´t it bizarre that a Spanish nurse had a chance of survival, while there´s no cure for Africans left to die in horrendous circumstances?
If Dr Broderick, Dr Horowitz and Shimatsu´s research proves correct, then this could be the most heinous crime in the history of humanity. It also means that Ebola screening at airports, military presence in Africa, and tougher legislation on immigration could be nothing more than a convenient reason to further destroy our civil liberties in the name of security- and cause more division, fear and hatred between different nations and ethnic groups.
http://rense.com/general96/ebobreakout.html

Tämän USA:n omistaman patentin numero on CA2741523A1 ja katseltavissa alla olevasta linkistä.
http://www.google.com/patents/CA2741523A1
http://www.liberianobserver.com/security/ebola-aids-manufactured-western-pharmaceuticals-us-dod
| Julkaisun numero | CA2741523 A1 |
| Julkaisutyyppi | Hakemus |
| Hakemusnumero | CA 2741523 |
| PCT-numero | PCT/US2009/062079 |
| Julkaisupäivämäärä | 29. huhtikuu 2010 |
| Hakemuksen jättöpäivä | 26. lokakuu 2009 |
| Etuoikeuspäivä | 24. lokakuu 2008 |
| Julkaistu lisäksi otsikolla | EP2350270A2, EP2350270A4,US20120251502, WO2010048615A2,WO2010048615A3, Vähemmän « |
| Keksijät | Jonathan S. Towner, Stuart T. Nichol, James A. Comer, Thomas G. Ksiazek, Pierre E. Rollin, Vähemmän « |
| Hakija | Jonathan S. Towner, Stuart T. Nichol, James A. Comer, Thomas G. Ksiazek, Pierre E. Rollin, The Government Of The United States Of America As Represented By The Sec Retary, Department Of Health & Human Services, Center For Disease Contro,Vähemmän « |
| Vie sitaatti | BiBTeX, EndNote, RefMan |
| Luokittelu (21), Oikeudelliset tapahtumat (1) | |
| Ulkoiset linkit: CIPO, Espacenet | |
Human ebola virus species and compositions and methods thereof
CA 2741523 A1
CA 2741523 A1
TIIVISTELMÄ
Compositions and methods including and related to the Ebola Bundibugyo virus (EboBun) are provided.
Compositions are provided that are operable as immunogens to elicit and immune response or protection from EboBun challenge in a subject such as a primate. Inventive methods are directed to detection and treatment of EboBun infection.
Compositions are provided that are operable as immunogens to elicit and immune response or protection from EboBun challenge in a subject such as a primate. Inventive methods are directed to detection and treatment of EboBun infection.
VAATIMUKSET(30)
1. An isolated hEbola virus comprising a nucleic acid molecule comprising a nucleotide sequence of:
a) a nucleotide sequence set forth in SEQ ID NOS: 1 or 10;
b) a nucleotide sequence hybridizing under stringent conditions to SEQ ID NOS:
1 or 10; or c) a nucleotide sequence of at least 70%-99% identity to the SEQ ID NOS: 1 or 10.
a) a nucleotide sequence set forth in SEQ ID NOS: 1 or 10;
b) a nucleotide sequence hybridizing under stringent conditions to SEQ ID NOS:
1 or 10; or c) a nucleotide sequence of at least 70%-99% identity to the SEQ ID NOS: 1 or 10.
2. An isolated hEbola virus having Centers for Disease Control Deposit Accession No.
200706291.
200706291.
3. The hEbola virus of any one of claims 1 or 2 which is killed.
4. The hEbola virus of claim 1 which is an attenuated hEbola virus.
5. The virus of claim 4 wherein at least one property of the attenuated hEbola virus is reduced from among infectivity, replication ability, protein synthesis ability, assembling ability or cytopathic effect.
6. An isolated nucleic acid molecule comprising the nucleotide sequence of SEQ
ID
NOS: 1 or 10 or a complement thereof.
ID
NOS: 1 or 10 or a complement thereof.
7. An isolated nucleic acid molecule comprising a nucleotide sequence of between 4 and 4900 contiguous nucleotides of the nucleotide sequence of SEQ ID NOS: 1 or 10, or a complement thereof; with the proviso that said nucleotide sequence is not comprised by the nucleotide sequence set forth in SEQ ID NO: 20; or between 5500 and 6600 contiguous nucleotides of the nucleotide sequence of SEQ ID NOS: 1 or 10, or a complement thereof.
8. An isolated nucleic acid molecule comprising a nucleotide sequence that encodes the amino acid sequence of SEQ ID NO: 2-9, 59, or SEQ ID NO: 11-19 or a complement of said nucleotide sequence.
9. An isolated RNA or DNA nucleic acid molecule which hybridizes under stringent conditions to a nucleic acid molecule having the nucleotide sequence of SEQ ID
NOS: 1 or 10 or a complement thereof.
NOS: 1 or 10 or a complement thereof.
10. An isolated polypeptide encoded by the nucleic acid molecule of any one of claims 7-9.
11. An isolated polypeptide comprising the amino acid of:
a) an amino acid sequence set forth in any of SEQ ID NOS: 2-19, or 59; or b) an amino acid sequence that has 70% - 99% homology to the amino acid sequence of (a).
a) an amino acid sequence set forth in any of SEQ ID NOS: 2-19, or 59; or b) an amino acid sequence that has 70% - 99% homology to the amino acid sequence of (a).
12. An isolated polypeptide comprising the amino acid sequence having to 250 contiguous amino acid residues of the amino acid sequence of SEQ ID
NOS: 5 or 18 (VP24);
5 to 280 contiguous residues of the amino acid sequence of SEQ ID NOS: 6 or 17 (VP30);
5 to 320 contiguous residues of the amino acid sequence of SEQ ID NOS: 8 or 13 (VP40);
5 to 340 contiguous residues of the amino acid sequence of SEQ ID NOS: 7 or 12 (VP35);
5 to 370 contiguous residues of the amino acid sequence of SEQ ID NOS: 4 or 15 (SGP);
5 to 370 contiguous residues of the amino acid sequence of SEQ ID NOS: 59 or 16 (SSGP);
5 to 670 contiguous residues of the amino acid sequence of SEQ ID NOS: 9 or 14 (GP);
5 to 730 contiguous residues of the amino acid sequence of SEQ ID NOS: 3 or 11 (NP); or 5 to 2200 contiguous residues of the amino acid sequence of SEQ ID NOS: 2 or 19 (L).
NOS: 5 or 18 (VP24);
5 to 280 contiguous residues of the amino acid sequence of SEQ ID NOS: 6 or 17 (VP30);
5 to 320 contiguous residues of the amino acid sequence of SEQ ID NOS: 8 or 13 (VP40);
5 to 340 contiguous residues of the amino acid sequence of SEQ ID NOS: 7 or 12 (VP35);
5 to 370 contiguous residues of the amino acid sequence of SEQ ID NOS: 4 or 15 (SGP);
5 to 370 contiguous residues of the amino acid sequence of SEQ ID NOS: 59 or 16 (SSGP);
5 to 670 contiguous residues of the amino acid sequence of SEQ ID NOS: 9 or 14 (GP);
5 to 730 contiguous residues of the amino acid sequence of SEQ ID NOS: 3 or 11 (NP); or 5 to 2200 contiguous residues of the amino acid sequence of SEQ ID NOS: 2 or 19 (L).
13. An isolated antibody or an antigen-binding fragment thereof which immunospecifically binds to the hEbola virus of any one of claims 1or 2 or neutralizes the virus.
14. An isolated antibody or an antigen-binding fragment thereof which immunospecifically binds to the polypeptide of any one of claims 11 or 12 or neutralizes an hEbola virus.
15. A method for detecting the presence of a the hEbola virus or a nucleic acid molecule derived therefrom of claim 1 in a biological sample, said method comprising:
(a) contacting the sample with an agent that selectively binds to the virus or the nucleic acid molecule derived therefrom; and (b) detecting whether the compound binds to the virus or the nucleic acid molecule derived therefrom in the sample.
(a) contacting the sample with an agent that selectively binds to the virus or the nucleic acid molecule derived therefrom; and (b) detecting whether the compound binds to the virus or the nucleic acid molecule derived therefrom in the sample.
16. The method of claim 15, wherein the agent is an antibody.
17. The method of claim 15, wherein the agent is a nucleic acid molecule comprising a nucleotide sequence having between 4 and 6600 contiguous nucleotides of the nucleotide sequence of SEQ ID NOS: 1 or 10, or a complement thereof.
18. A method for detecting the presence of the polypeptide of claim 11 in a biological sample, said method comprising:
(a) contacting the biological sample with an agent that selectively binds to said polypeptide; and (b) detecting whether the compound binds to said polypeptide in the sample.
(a) contacting the biological sample with an agent that selectively binds to said polypeptide; and (b) detecting whether the compound binds to said polypeptide in the sample.
19. The method of claim 18, wherein the agent is an antibody or an antigen-binding fragment thereof.
20. A formulation comprising the hEbola virus of any one of claims 3 or 4, and a pharmaceutically acceptable carrier.
21. A formulation comprising an amount of a protein extract of the hEbola virus of claim 3 or 4 or a subunit thereof, and a pharmaceutically acceptable carrier.
22. A formulation comprising an amount of a nucleic acid molecule of the nucleotide sequence of SEQ ID NOS: 1 or 10 or a complement thereof, and a pharmaceutically acceptable carrier.
23. A formulation comprising an immunogenically effective amount of the nucleic acid molecule of claim 9 or a complement thereof, and a pharmaceutically acceptable carrier.
24. A vaccine formulation comprising a therapeutically or prophylactically effective amount of the hEbola virus of claim 3 or 4 or a protein extract therefrom, and a pharmaceutically acceptable carrier.
25. A vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule SEQ ID NOS: 1 or 10 or a complement thereof, and a pharmaceutically acceptable carrier.
26. A vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule of claim 9 or a complement thereof, and a pharmaceutically acceptable carrier.
27. A pharmaceutical composition comprising a prophylactically or therapeutically effective amount of an anti-hEbola agent of an antibody or an antigen-binding fragment thereof which immunospecifically binds to the hEbola virus of Deposit Accession No.
200706291, or polypeptides or protein derived therefrom and optionally has the nucleotide sequence of SEQ ID
NOS: 1 or 10, or a fragment thereof.
200706291, or polypeptides or protein derived therefrom and optionally has the nucleotide sequence of SEQ ID
NOS: 1 or 10, or a fragment thereof.
28. A kit comprising a container containing the formulation of any one of claims 24-26.
29. A method for identifying a subject infected with the virus of claim 1 or 2, comprising:
(a) obtaining total RNA from a biological sample obtained from the subject;
(b) reverse transcribing the total RNA to obtain cDNA; and (c) amplifying the cDNA using a set of primers derived from a nucleotide sequence of the virus of claim 1 or 2.
(a) obtaining total RNA from a biological sample obtained from the subject;
(b) reverse transcribing the total RNA to obtain cDNA; and (c) amplifying the cDNA using a set of primers derived from a nucleotide sequence of the virus of claim 1 or 2.
30. A primer that has the nucleotide sequence of one of SEQ ID NOS: 24-57.
KUVAUS (OCR-teksti voi sisältää virheitä)
HUMAN EBOLA VIRUS SPECIES AND COMPOSITIONS AND METHODS THEREOF
DEPOSIT STATEMENT
[0001] The invention provides the isolated human Ebola (hEbola) viruses denoted as Bundibugyo (EboBun) deposited with the Centers for Disease Control and Prevention ("CDC";
Atlanta, Georgia, United States of America) on November 26, 2007 and accorded an accession number 200706291. This deposit was not made to an International Depository Authority (IDA) as established under the Budapest Treaty on the International Recognition of the Deposit of Microorganisms for the Purposes of Patent Procedure, and is a non-Budapest treaty deposit. The deposited organism is not acceptable by American Type Culture Collection (ATCC), Manassas, Virginia, an International Depository Authority (IDA) as established under the Budapest Treaty on the International Recognition of the Deposit of Microorganisms for the Purposes of Patent Procedure. Samples of the stated Deposit Accession No. 200706291 will be made available to approved facilities for thirty years from the date of deposit, and for the lifetime of the patent issuing from, or claiming priority to this application.
RELATED APPLICATIONS
[0002] This application claims priority benefit of U.S. Provisional Application 61/108,175 filed 24 October 2008; the contents of which are hereby incorporated by reference.
FIELD OF THE INVENTION
[0003] The invention is related to compositions and methods directed to a novel species of human Ebola (hEbola) virus.
BACKGROUND OF THE INVENTION
[0004] The family Filoviridae consists of two genera, Marburgvirus and Ebolavirus, which have likely evolved from a common ancestor'. The genus Ebolavirus includes four species: Zaire, Sudan, Reston and Cote d'Ivoire (Ivory Coast) ebolaviruses, which have, with the exception of Reston and Cote d'Ivoire ebolaviruses, been associated with large hemorrhagic fever (HF) outbreaks in Africa with high case fatality (53-90%)2.
[0005] Viruses of each species have genomes that are at least 30-40% divergent from one another, a level of diversity that presumably reflects differences in the ecologic niche they occupy and in their evolutionary history. Identification of the natural reservoir of ebolaviruses remains somewhat elusive, although recent PCR and antibody data suggest that three species of arboreal fruit bats may be carriers of Zaire ebolavirus3. No data has yet been published to suggest reservoirs for the Sudan, Reston and Cote d'Ivoire ebolavirus species. However, a cave-dwelling fruit bat has been recently implicated as a natural host for marburgvirus4' s, supporting the hypothesis that different bat species may be the reservoir hosts for the various filoviruses.
[0006] Filovirus outbreaks are sporadic, sometimes interspersed by years or even decades of no apparent disease activity. The last new species of ebolavirus was discovered 14 years ago (1994), in Cote d'Ivoire (Ivory Coast), and involved a single non-fatal case, a veterinarian who performed an autopsy on an infected chimpanzee found in the Tai Forest6. No further disease reports have been associated with Cote d'Ivoire ebolavirus, in contrast to Zaire and Sudan ebolaviruses which have each caused multiple large outbreaks over the same time period.
[0007] In late November 2007, HF cases were reported in the townships of Bundibugyo and Kikyo in Bundibugyo District, Western Uganda. The outbreak continued through January 2008, and resulted in approximately 149 cases and 37 deaths. Laboratory investigation of the initial 29 suspect-case blood specimens by classic methods (antigen capture, IgM and IgG
ELISA) and a recently developed random-primed pyrosequencing approach identified this to be an Ebola HF
outbreak associated with a new discovered ebolavirus species. These specimens were negative when initially tested with highly sensitive real-time RT-PCR assays specific for all known Zaire and Sudan ebolaviruses and Marburg viruses. This new species is referred to herein as "the Bundibugyo species", abbreviated "EboBun".
[0008] Accordingly, compositions and methods directed to the new Ebola virus species are described herein and the most closely related Ebola Ivory Coast species, which compositions and methods are useful for diagnosis and prevention of human Ebola virus infection; including related vaccine development, and prevention of hemorrhagic fever in a human population.
SUMMARY OF THE INVENTION
[0009] The present invention is based upon the isolation and identification of a new human Ebola virus species, EboBun. EboBun was isolated from the patients suffering from hemorrhagic fever in a recent outbreak in Uganda. The isolated virus is a member of the Filoviridae family, a family of negative sense RNA viruses. Accordingly, the invention relates to the isolated EboBun virus that morphologically and phylogenetically relates to known members filoviridae.
[0010] In one aspect, the invention provides the isolated EboBun virus deposited with the Centers for Disease Control and Prevention ("CDC"; Atlanta, Georgia, United States of America) on November 26, 2007 and accorded an accession number 200706291, as stated in the paragraph entitled "DEPOSIT STATEMENT" supra.
[0011] In another aspect, the invention provides an isolated hEbola EboBun virus comprising a nucleic acid molecule comprising a nucleotide sequence selected from the group consisting of: a) a nucleotide sequence set forth in SEQ ID NO: 1; b) a nucleotide sequence that hybridizes to the sequence set forth in SEQ ID NO: 1 under stringent conditions; and c) a nucleotide sequence that has at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to the SEQ ID NO:
1. In another aspect, the invention provides the complete genomic sequence of the hEbola virus EboBun.
[0012] In a related aspect, the invention provides nucleic acid molecules isolated from EboBun, or fragments thereof.
[0013] In another aspect, the invention provides proteins or polypeptides that are isolated from the EboBun, including viral proteins isolated from cells infected with the virus but not present in comparable uninfected cells; or fragments thereof. In one embodiment of the present invention, the amino acid sequences of the proteins or polypeptides are set forth in SEQ ID
NOS: 2-9 and 59, or fragments thereof.
[0014] In a related aspect, the invention provides an isolated polypeptide encoded by the nucleic acid molecule of the inventive hEbola EboIC (Sequence ID No. 10) virus described above.
[0015] In another aspect, the invention provides an isolated hEbola EboIC
virus comprising a nucleic acid molecule comprising a nucleotide sequence selected from the group consisting of: a) a nucleotide sequence set forth in SEQ ID NO: 10; b) a nucleotide sequence that hybridizes to the sequence set forth in SEQ ID NO: 10 under stringent conditions; and c) a nucleotide sequence that has at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to the SEQ ID NO:
10. In another aspect, the invention provides the complete genomic sequence of the hEbola virus EboIC.
[0016] In a related aspect, the invention provides nucleic acid molecules isolated from EboIC, or fragments thereof.
[0017] In another aspect, the invention provides proteins or polypeptides that are isolated from the EboIC, including viral proteins isolated from cells infected with the virus but not present in comparable uninfected cells; or fragments thereof. In one embodiment of the present invention, the amino acid sequences of the proteins or polypeptides are set forth in SEQ ID
NOs: 11-19, or fragments thereof.
[0018] In a related aspect, the invention provides an isolated polypeptide encoded by the nucleic acid molecule of the inventive hEbola EboIC virus described above.
[0019] In other aspects, the invention relates to the use of the isolated hEbola virus for diagnostic and therapeutic methods based on EbBun, EboIC, or a combination thereof. In one embodiment, the invention provides a method of detecting in a biological sample an antibody immunospecific for the genus of West Afrin Ebola Species constituting hEbola EbBun and EboIC
virus using at least one the inventive isolated hEbola virus described herein, or any of the inventive proteins or polypeptides as described herein. In another specific embodiment, the invention provides a method of screening for an antibody which immunospecifically binds and neutralizes hEbola EboBun. Such an antibody is useful for a passive immunization or immunotherapy of a subject infected with hEbola.
[0020] In another aspect, the invention provides an isolated antibody or an antigen-binding fragment thereof which immunospecifically binds to the hEbola virus of the invention described above.
[0021] In other aspects, the invention provides methods for detecting the presence, activity or expression of the Glade of Bundibungyo-Ivory Coast hEbola virus in a biological material, such as cells, blood, saliva, urine, feces and so forth; and specifically at least one of EbBun or EboIC.
[0022] In a related aspect, the invention provides a method for detecting the presence of the inventive hEbola virus described above in a biological sample, the method includes (a) contacting the sample with an agent that selectively binds to a West African hEbola virus; and (b) detecting whether the compound binds to the West African hEbola virus in the sample.
[0023] In another aspect, the invention provides a method for detecting the presence of the inventive polypeptide described above, in a biological sample, said method includes (a) contacting the biological sample with an agent that selectively binds to the polypeptide;
and (b) detecting whether the agent binds to the polypeptide in the sample. In another aspect, the invention provides a method for detecting the presence of a first nucleic acid molecule derived from the inventive hEbola virus described above in a biological sample, the method comprising: (a) contacting the biological sample with an agent that selectively binds to the polypeptide; and (b) detecting whether the agent binds to the polypeptide in the sample.
[0024] In another aspect, the invention provides a method for propagating the hEbola virus in host cells comprising infecting the host cells with the inventive isolated hEbola virus described above, culturing the host cells to allow the virus to multiply, and harvesting the resulting virions.
Also provided by the present invention are host cells infected with the inventive hEbola virus 5 described above.
[0025] In another aspect, the invention provides a method of detecting in a biological sample the presence of an antibody that immunospecifically binds hEbola virus, the method comprising: (a) contacting the biological sample with the inventive host cell host described above; and (b) detecting the antibody bound to the cell.
[0026] In another aspect, the invention provides vaccine preparations, comprising the inventive hEbola virus, including recombinant and chimeric forms of the virus, nucleic acid molecules comprised by the virus, or protein subunits of the virus. The invention also provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of the inventive hEbola virus described above, and a pharmaceutically acceptable carrier. In one embodiment, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a protein extract of the inventive hEbola virus described above, or a subunit thereof; and a pharmaceutically acceptable carrier. In another, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NO: 1 or a complement thereof, and a pharmaceutically acceptable carrier. In another, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule comprising any of inventive the nucleotide sequences as described above, or a complement thereof, and a pharmaceutically acceptable carrier.
[0027] In a related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of the inventive hEbola virus described above, and a pharmaceutically acceptable carrier. In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of a protein extract of the inventive hEbola virus described above or a subunit thereof, and a pharmaceutically acceptable carrier. In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NO: 1 or a complement thereof, and a pharmaceutically acceptable carrier. In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of a nucleic acid molecule comprising the inventive nucleotide sequence as described above or a complement thereof, and a pharmaceutically acceptable carrier. In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of any of the inventive polypeptides described above.
[0028] In another aspect, the present invention provides pharmaceutical compositions comprising antiviral agents of the present invention and a pharmaceutically acceptable carrier. In a specific embodiment, the antiviral agent of the invention is an antibody that immunospecifically binds hEbola virus or any hEbola epitope. In another specific embodiment, the antiviral agent is a polypeptide or protein of the present invention or nucleic acid molecule of the invention.
[0029] In a related aspect, the invention provides a pharmaceutical composition comprising a prophylactically or therapeutically effective amount of an anti-hEbola EboBun agent and a pharmaceutically acceptable carrier.
[0030] The invention also provides kits containing compositions and formulations of the present invention. Thus, in another aspect, the invention provides a kit comprising a container containing the inventive immunogenic formulation described above. In another aspect, the invention provides a kit comprising a container containing the inventive vaccine formulation described above. In another, the invention provides a kit comprising a container containing the inventive pharmaceutical composition described above. In another, the invention provides a kit comprising a container containing the inventive vaccine formulation described above. In another, the invention provides a method for identifying a subject infected with the inventive hEbola virus described above, comprising: (a) obtaining total RNA from a biological sample obtained from the subject; (b) reverse transcribing the total RNA to obtain cDNA; and (c) amplifying the cDNA using a set of primers derived from a nucleotide sequence of the inventive hEbola virus described above.
[0031] The invention further relates to the use of the sequence information of the isolated virus for diagnostic and therapeutic methods.
[0032] In another aspect, the present invention provides methods for screening antiviral agents that inhibit the infectivity or replication of hEbola virus or variants thereof.
[0033] The invention further provides methods of preparing recombinant or chimeric forms of hEbola.
BRIEF DESCRIPTION OF THE DRAWINGS
[0034] FIG. 1 represents a Phylogenetic tree comparing full-length genomes of Ebolavirus and Marburg virus by Bayesian analysis;
[0035] FIG. 2 represents an alignment of genomes of novel hEbola EboBun (SEQ
ID NO: 1) referred to below as "Ebola Bundibugyo" or "EboBun", and hEbola Zaire (SEQ ID
NO: 20);
referred to below as "Ebola Zaire `76" or "EboZ" and hEbola Ivory Coast (SEQ
ID NO: 10) also referred to below as "EboIC".
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
[0036] It is to be understood that the present invention is not limited to particular embodiments described, as such may, of course, vary. It is also to be understood that the terminology used herein is for the purpose of describing particular embodiments only, and is not intended to be limiting.
[0037] Due to the sequence divergence of EboBun relative to all previously recognized ebolaviruses, the present invention has utility in design of diagnostic assays to monitor Ebola HF
disease in humans and animals, and develop effective antivirals and vaccines.
[0038] The EboBun virus of the present invention is genetically distinct, differing by more than 30% at the genome level from all other known ebolavirus species. The unique nature of this virus created challenges for traditional filovirus molecular based diagnostic assays and genome sequencing approaches. Instead, over 70% of the virus genome was sequenced using a recently developed random-primed pyrosequencing approach which allowed the rapid development of molecular detection assay which were deployed in the disease outbreak response. This random-primed pyrosequencing draft sequence allowed faster completion of the whole genome sequence using traditional primer walking approach and confirmation that the EboBun virus represented a new ebolavirus species.
Definitions [0039] The definitions herein provided are operative throughout the entire description of the invention set forth herein, including the Summary of the Invention.
[0040] The term "an antibody or an antibody fragment that immunospecifically binds a polypeptide of the invention" as used herein refers to an antibody or a fragment thereof that immunospecifically binds to the polypeptide encoded by the nucleotide sequence of SEQ ID NO: 1 (EboBun), or a fragment thereof, and does not non-specifically bind to other polypeptides. An antibody or a fragment thereof that immunospecifically binds to the polypeptide of the invention may cross-react with other antigens. Preferably, an antibody or a fragment thereof that immunospecifically binds to a polypeptide of the invention does not cross-react with other antigens.
An antibody or a fragment thereof that immunospecifically binds to the polypeptide of the invention can be identified by, for example, immunoassays or other techniques known to those skilled in the art, or otherwise as described herein.
[0041] An "isolated" or "purified" peptide or protein is substantially free of cellular material or other contaminating proteins from the cell or tissue source from which the protein is derived, or substantially free of chemical precursors or other chemicals when chemically synthesized. The language "substantially free of cellular material" includes preparations of a polypeptide/protein in which the polypeptide/protein is separated from cellular components of the cells from which it is isolated or recombinantly produced. Thus, a polypeptide/protein that is substantially free of cellular material includes preparations of the polypeptide/protein having less than about 30%, 20%, 10%, 5%, 2.5%, or 1% (by dry weight) of contaminating protein. When the polypeptide/protein is recombinantly produced, it is also preferably substantially free of culture medium, i.e., culture medium represents less than about 20%, 10%, or 5% of the volume of the protein preparation.
When polypeptide/protein is produced by chemical synthesis, it is preferably substantially free of chemical precursors or other chemicals, i.e., it is separated from chemical precursors or other chemicals which are involved in the synthesis of the protein. Accordingly, such preparations of the polypeptide/protein have less than about 30%, 20%, 10%, 5% (by dry weight) of chemical precursors or compounds other than polypeptide/protein fragment of interest.
In a preferred embodiment of the present invention, polypeptides/proteins are isolated or purified.
[0042] An "isolated" nucleic acid molecule is one which is separated from other nucleic acid molecules which are present in the natural source of the nucleic acid molecule. Moreover, an "isolated" nucleic acid molecule, such as a cDNA molecule, can be substantially free of other cellular material, or culture medium when produced by recombinant techniques, or substantially free of chemical precursors or other chemicals when chemically synthesized. In a preferred embodiment of the invention, nucleic acid molecules encoding polypeptides/proteins of the invention are isolated or purified. The term "isolated" nucleic acid molecule does not include a nucleic acid that is a member of a library that has not been purified away from other library clones containing other nucleic acid molecules.
[0043] The term "portion" or "fragment" as used herein includes the specified fragment lengths, and all integers in between, inclusive of the specified end points in a specified range, and inclusive of any length up to the full length of a protein, polypeptide, or nucleic acid.
[0044] The term "having a biological activity of the protein" or "having biological activities of the polypeptides of the invention" refers to the characteristics of the polypeptides or proteins having a common biological activity, similar or identical structural domain, and/or having sufficient amino acid identity to the polypeptide encoded by the nucleotide sequence of SEQ ID
NO: 1 (EboBun).
Such common biological activities of the polypeptides of the invention include antigenicity and immunogenicity.
[0045] The term "under stringent condition" refers to hybridization and washing conditions under which nucleotide sequences having at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, or at least 95% identity to each other remain hybridized to each other. Such hybridization conditions are described in, for example but not limited to, Current Protocols in Molecular Biology, John Wiley & Sons, NY (1989), 6.3.1-6.3.6.; Basic Methods in Molecular Biology, Elsevier Science Publishing Co., Inc., NY (1986), pp. 75-78, and 84-87; and Molecular Cloning, Cold Spring Harbor Laboratory, NY (1982), pp. 387-389, and are well known to those skilled in the art. A preferred, non-limiting example of stringent hybridization conditions is hybridization in 6 x sodium chloride/sodium citrate (SSC), 0.5% SDS at about 68 C followed by one or more washes in 2 x SSC, 0.5% SDS at room temperature. Another preferred, non-limiting example of stringent hybridization conditions is hybridization in 6 x SSC at about 45 C, followed by one or more washes in 0.2 x SSC, 0.1% SDS at about 50-65 C.
[0046] The term "variant" as used herein refers either to a naturally occurring genetic mutant of hEbola EboBun, or hEbola EboIC, or a recombinantly prepared variation of these hEbola species, each of which contain one or more mutations in its genome compared to the hEbola of SEQ ID NO:
1 or 10. The term "variant" may also refer either to a naturally occurring variation of a given peptide or a recombinantly prepared variation of a given peptide or protein in which one or more amino acid residues have been modified by amino acid substitution, addition, or deletion.
[0047] "Homology" refers to sequence similarity or, alternatively, sequence identity, between two or more polynucleotide sequences or two or more polypeptide sequences.
[0048] The terms "percent identity" and "% identity," as applied to polynucleotide sequences, refer to the percentage of identical nucleotide matches between at least two polynucleotide sequences aligned using a standardized algorithm. Such an algorithm may insert, in a standardized and reproducible way, gaps in the sequences being compared in order to optimize alignment between two sequences, and therefore achieve a more meaningful comparison of the two sequences.
[0049] Percent identity between polynucleotide sequences may be determined using one or more computer algorithms or programs known in the art or described herein. For example, percent 5 identity can be determined using the default parameters of the CLUSTAL V
algorithm as incorporated into the MEGALIGN version 3.12e sequence alignment program. This program is part of the LASERGENE software package, a suite of molecular biological analysis programs (DNASTAR, Madison, Wis.). CLUSTAL V is described in Higgins, D. G. and P. M.
Sharp (1989;
CABIOS 5:151-153) and in Higgins, D. G. et al. (1992; CABIOS 8:189-191). For pairwise 10 alignments of polynucleotide sequences, the default parameters are set as follows: Ktuple=2, gap penalty=5, window=4, and "diagonals saved"=4. The "weighted" residue weight table is selected as the default.
[0050] Alternatively, a suite of commonly used and freely available sequence comparison algorithms which can be used is provided by the National Center for Biotechnology Information (NCBI) Basic Local Alignment Search Tool (BLAST) (Altschul, S. F. et al.
(1990) J. Mol. Biol.
215:403-410), which is available from several sources, including the NCBI, Bethesda, Md., and on the NCBI World Wide Web site available on the Internet. The BLAST software suite includes various sequence analysis programs including "blastn," that is used to align a known polynucleotide sequence with other polynucleotide sequences from a variety of databases. Also available is a tool called "BLAST 2 Sequences" that is used for direct pairwise comparison of two nucleotide sequences. "BLAST 2 Sequences" can be accessed and used interactively on the Internet via the NCBI World Wide Web site as well. The "BLAST 2 Sequences" tool can be used for both blastn and blastp (discussed below). BLAST programs are commonly used with gap and other parameters set to default settings. For example, to compare two nucleotide sequences, one may use blastn with the "BLAST 2 Sequences" tool Version 2Ø12 (Apr. 21, 2000) set at default parameters. Such default parameters may be, for example: Matrix:BLOSUM62; Reward for match: 1;
Penalty for mismatch: -2; Open Gap: 5 and Extension Gap: 2 penalties; Gap x drop-off: 50;
Expect: 10; Word Size: 11; Filter: on.
[0051] Percent identity may be measured over the length of an entire defined sequence, for example, as defined by a particular SEQ ID number, or may be measured over a shorter length, for example, over the length of a fragment taken from a larger, defined sequence, for instance, a fragment of at least 20, at least 30, at least 40, at least 50, at least 70, at least 100, or at least 200 contiguous nucleotides. Such lengths are exemplary only, and it is understood that any fragment length supported by the sequences shown herein, in the tables, figures, or sequence listing, may be used to describe a length over which percentage identity may be measured.
[0052] The phrases "percent identity" and "% identity", as applied to polypeptide sequences, refer to the percentage of identical residue matches between at least two polypeptide sequences aligned using a standardized algorithm. Methods of polypeptide sequence alignment are well known. Some alignment methods take into account conservative amino acid substitutions. Such conservative substitutions, explained in more detail above, generally preserve the charge and hydrophobicity at the site of substitution, thus preserving the structure (and therefore function) of the polypeptide. The phrases "percent similarity" and "% similarity", as applied to polypeptide sequences, refer to the percentage of residue matches, including identical residue matches and conservative substitutions, between at least two polypeptide sequences aligned using a standardized algorithm. In contrast, conservative substitutions are not included in the calculation of percent identity between polypeptide sequences.
[0053] Percent identity between polypeptide sequences may be determined using the default parameters of the CLUSTAL V algorithm as incorporated into the MEGALIGN
version 3.12e sequence alignment program (described and referenced above). For pairwise alignments of polypeptide sequences using CLUSTAL V, the default parameters are set as follows: Ktuple=l, gap penalty=3, window=5, and "diagonals saved"=5. The PAM250 matrix is selected as the default residue weight table.
[0054] Alternatively the NCBI BLAST software suite may be used. For example, for a pairwise comparison of two polypeptide sequences, one may use the "BLAST 2 Sequences"
tool Version 2Ø12 (Apr. 21, 2000) with blastp set at default parameters. Such default parameters may be, for example: Matrix: BLOSUM62; Open Gap: 11 and Extension Gap: 1 penalties; Gap x drop-off: 50;
Expect: 10; Word Size: 3; Filter: on.
[0055] Percent identity may be measured over the length of an entire defined polypeptide sequence, for example, as defined by a particular SEQ ID number, or may be measured over a shorter length, for example, over the length of a fragment taken from a larger, defined polypeptide sequence, for instance, a fragment of at least 15, at least 20, at least 30, at least 40, at least 50, at least 70 or at least 150 contiguous residues. Such lengths are exemplary only, and it is understood that any fragment length supported by the sequences shown herein, in the tables, figures or sequence listing, may be used to describe a length over which percentage identity may be measured.
[0056] The term "agent" encompasses any chemical, biochemical, or biological molecule; such as small molecules, proteins, polypeptides, antibodies, nucleic acid molecules including DNA or RNA, and the like.
Methods and compositions related to the inventive hEbola [0057] The present invention is based upon the isolation and identification of a new human Ebola virus species, EboBun and the sequencing of the only other known West African Ebola species EboIC. EboBun was isolated from the patients suffering from hemorrhagic fever in a recent outbreak in Uganda. The isolated virus is a member of the Filoviridae family, a family of negative sense RNA viruses. Accordingly, the invention relates to the isolated EboBun or EBOIC virus that morphologically and phylogenetically relates to known members filoviridae.
[0058] In another aspect, the invention provides an isolated hEbola virus including a nucleic acid molecule with a nucleotide sequence that is preferably: a) a nucleotide sequence set forth in SEQ ID NO: 1; b) a nucleotide sequence that hybridizes to the sequence set forth in SEQ ID NO: 1 under stringent conditions; or c) a nucleotide sequence that has at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to the SEQ ID NO: 1. In one embodiment of the present invention, the hEbola virus is killed. In another, the virus is attenuated. In another, the infectivity of the attenuated hEbola virus is reduced. In another, the infectivity is reduced by at least 5-fold, 10-fold, 25-fold, 50-fold, 100-fold, 250-fold, 500-fold, or 10,000-fold. In another, the replication ability of the attenuated hEbola virus is reduced. In another, the replication ability of the attenuated virus is educed by at least 5-fold, 10-fold, 25-fold, 50-fold, 100-fold, 250-fold, 500-fold, 1,000-fold, or 10,000-fold. In another, the protein synthesis ability of the attenuated virus is reduced. In another, the protein synthesis ability is reduced by at least 5-fold, 10-fold, 25-fold, 50-fold, 100-fold, 250-fold, 500-fold, 1,000-fold, or 10,000-fold. In another, the assembling ability of the attenuated hEbola virus is reduced. In another, the assembling ability of the attenuated virus is reduced by at least 5-fold, 10-fold, 25-fold, 50-fold, 100-fold, 250-fold, 500-fold, 1,000-fold, or 10,000-fold. In another, the cytopathic effect of the attenuated hEbola virus is reduced. In another, the cytopathic effect is reduced by at least 5-fold, 10-fold, 25-fold, 50-fold, 100-fold, 250-fold, 500-fold, 1,000-fold, or 10,000-fold.
[0059] In another aspect, the invention provides the complete genomic sequence of the hEbola virus EboBun or EboIC. In a specific embodiment, the virus includes a nucleotide sequence of SEQ
ID NOs: 1 or 10, respectively.
[0060] In a related aspect, the invention provides nucleic acid molecules isolated from EboBun, EboIC, or fragments thereof. In one embodiment of the present invention, the isolated nucleic acid molecule includes the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof. In another, the nucleic acid molecule includes a nucleotide sequence having at least 4, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 4600, 4700, 4800, or 4900 contiguous nucleotides of the nucleotide sequence of SEQ ID NO: 1, or a complement thereof;
with the proviso that the nucleotide sequence is not comprised by the nucleotide sequence set forth in SEQ ID NO: 20 (Ebola Zaire nucleotide sequence); or at least 5000, 5500, 5600, 5700, 5800, 5900, 6000, 6100, 6200, 6300, 6400, 6500, or 6600 contiguous nucleotides of the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof. In another embodiment, the isolated nucleic acid molecule includes a nucleotide sequence that encodes the EboBun amino acid sequence of SEQ ID NOs: 2-9 or 59, the EboIC amino acid sequence of SEQ ID NOs: 11-19, or a complement of the nucleotide sequence that encodes the EboBun amino acid sequences of SEQ
ID NOs: 2-9 or 59 or the EboIC amino acid sequences of SEQ ID NOs: 11-19. In another, the isolated nucleic acid molecule hybridizes under stringent conditions to a nucleic acid molecule having the nucleotide sequence of SEQ ID NOs: 1 or 10 or a complement thereof, wherein the nucleic acid molecule encodes an amino acid sequence which has a biological activity exhibited by a polypeptide encoded by the nucleotide sequence of SEQ ID NOs: 1 or 10. In another, nucleic acid molecule is RNA. In another, nucleic acid molecule is DNA.
[0061] In another aspect, the invention provides proteins or polypeptides that are isolated from the EboBun, including viral proteins isolated from cells infected with the virus but not present in comparable uninfected cells. In one embodiment of the present invention, the amino acid sequences of the proteins or polypeptides are set forth in SEQ ID NOs: 2-9, 59, or 11-19, or fragments thereof.
In one embodiment, polypeptides or proteins of the present invention have a biological activity of the protein (including antigenicity and/or immunogenicity) encoded by the sequence of SEQ ID
NOs: 1 or 10. In another, the polypeptides or the proteins of the present invention have a biological activity of at least one protein having the amino acid sequence (including antigenicity and/or immunogenicity) set forth in SEQ ID NOS: 2-9, 59, or 11-19, or a fragment thereof.
[0062] In a related aspect, the invention provides an isolated polypeptide encoded by the nucleic acid molecule of the invention described above. In one embodiment of the present invention, the isolated polypeptide includes the amino acid sequence selected from the group consisting of: a) an amino acid sequence set forth in SEQ ID NO: 2, 3, 4, 5, 6, 7, 8, or 9; 11, 12, 13, 14, 15, 16, 17, 18 or 19; and b) an amino acid sequence that has 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% homology to the amino acid sequence according to a). In another, the isolated polypeptide comprises the amino acid sequence having at least 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 210, 220, 230, 240 or 250 contiguous amino acid residues of the amino acid sequence of SEQ ID NOs: 5 or 18 (VP24); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 210, 220, 230, 240, 250, 260, 270, 280 contiguous amino acid residues of the amino acid sequence of SEQ ID NOs: 6 or 17 (VP30); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 310, or 320 contiguous amino acid residues of the amino acid sequence of SEQ
ID NOs: 8 or 13 (VP40); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 310, 320, 330, or 340 contiguous amino acid residues of the amino acid sequence of SEQ ID NOs: 7 or 12 (VP35); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 310, 320, 330, 340, 350, 360, or 370 contiguous amino acid residues of the amino acid sequence of SEQ ID
NOs: 4 or 15 (SGP); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 310, 320, 330, 340, 350, 360, or 370 contiguous amino acid residues of the amino acid sequence of SEQ ID NOs: 59 or 16 (SSGP); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, 450, 500, 550, 600, 610, 620, 630, 640, 650, 660, or 670 contiguous amino acid residues of the amino acid sequence of SEQ ID NOs: 9 or 14 (GP); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, 450, 500, 550, 600, 650, 700, 710, 720, or 730 contiguous amino acid residues of the amino acid sequence of SEQ
ID NOs: 3 or 11 (NP); or 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1050, 1100, 1150, 1200, 1250, 1300, 1350, 1400, 1450, 1500, 1550, 1600, 1650, 1700, 1750, 1800, 1850, 1900, 1950, 2000, 2050, 2100, 2150, 2160, 2170, 2180, 2190, or 2200 contiguous amino acid residues of the amino acid sequence of SEQ ID NOs: 2 or 19 (L).
[0063] In other aspects, the invention relates to the use of an isolated West African hEbola virus for diagnostic and therapeutic methods. In one embodiment, the invention provides a method of detecting in a biological sample an antibody immunospecific for the hEbola virus using the inventive isolated hEbola virus described herein, or any of the inventive proteins or polypeptides as described herein. In another specific embodiment, the invention provides a method of screening for an antibody which immunospecifically binds and neutralizes hEbola EboBun or EboIC
or a combination thereof. Such an antibody is useful for a passive immunization or immunotherapy of a subject infected with hEbola.
[0064] In another aspect, the invention provides an isolated antibody or an antigen-binding fragment thereof which immunospecifically binds to a West African genus hEbola virus of the 5 invention described above, and illustratively including EboBun or EboIC. In one embodiment of the present invention, the isolated antibody or an antigen-binding fragment thereof neutralizes a West African genus hEbola virus. In another, the isolated antibody or an antigen-binding fragment thereof immunospecifically binds to the inventive polypeptide described above. The invention further provides antibodies that specifically bind a polypeptide of the invention encoded by the nucleotide 10 sequence of SEQ ID NOs: 1 (EboBun) or 10 (EboIC), a fragment thereof, or encoded by a nucleic acid comprising a nucleotide sequence that hybridizes under stringent conditions to the nucleotide sequence of SEQ ID NOs: 1 (EboBun) or 10 (EboIC) and/or any hEbola EboBun epitope, having one or more biological activities of a polypeptide of the invention. These polypeptides include those shown in SEQ ID NOs: 2-9, 59, and 11-19. Such antibodies include, but are not limited to, 15 polyclonal, monoclonal, bi-specific, multi-specific, human, humanized, chimeric antibodies, single chain antibodies, Fab fragments, F(ab')2 fragments, disulfide-linked Fvs, intrabodies and fragments containing either a VL or VH domain or even a complementary determining region (CDR) that specifically binds to a polypeptide of the invention.
[0065] In other aspects, the invention provides methods for detecting the presence, activity or expression of the hEbola virus of the invention in a biological material, such as cells, blood, saliva, urine, and so forth. The increased or decreased activity or expression of the hEbola virus in a sample relative to a control sample can be determined by contacting the biological material with an agent which can detect directly or indirectly the presence, activity or expression of the hEbola virus.
In one embodiment of the present invention, the detecting agents are the antibodies or nucleic acid molecules of the present invention. Antibodies of the invention can also be used to treat hemorrhagic fever.
[0066] In a related aspect, the invention provides a method for detecting the presence of the inventive hEbola virus described above in a biological sample, the method comprising:
(a) contacting the sample with an agent that selectively binds to the hEbola virus; and (b) detecting whether the compound binds to the hEbola virus in the sample. In one embodiment of the present invention, the biological sample is selected from the group consisting of cells; blood; serum; plasma;
feces; rectal, vaginal and conjunctival swabs In another, the agent that binds to the virus is an antibody. In another, the agent that binds to the virus is a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NO: 1 or a complement thereof. In another, the agent that binds to the virus is a nucleic acid molecule comprising a nucleotide sequence having at least 4, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 4600, 4700, 4800, 4900, 5000, 5500, 5600, 5700, 5800, 5900, 6000, 6100, 6200, 6300, 6400, 6500, or 6600 contiguous nucleotides of the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof.
[0067] In another aspect, the invention provides a method for detecting the presence of the inventive polypeptide described above, in a biological sample, the method comprising:
(a) contacting the biological sample with an agent that selectively binds to the polypeptide; and (b) detecting whether the agent binds to the polypeptide in the sample. In one embodiment of the present invention, the biological sample is selected from the group consisting of cells; blood; serum;
plasma; feces; rectal, vaginal and conjunctival swabs. In another, the agent that binds to the polypeptide is an antibody or an antigen-binding fragment thereof.
[0068] In another aspect, the invention provides a method for detecting the presence of a first nucleic acid molecule derived from the inventive hEbola virus described above in a biological sample, the method includes (a) contacting the biological sample with an agent that selectively binds to the nucleic acid; and (b) detecting whether the agent binds to the nucleotide in the sample. In one embodiment of the present invention, the agent that binds to the first nucleic acid molecule is a second nucleic acid molecule comprising the nucleotide sequence of SEQ ID NO:
1 or a complement thereof. In another, the second nucleic acid molecule comprises at least 4, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 4600, 4700, 4800, 4900, 5000, 5500, 5600, 5700, 5800, 5900, 6000, 6100, 6200, 6300, 6400, 6500, or 6600 contiguous nucleotides of the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof.
[0069] In another aspect, the invention provides a method for propagating the hEbola virus in host cells comprising infecting the host cells with an inventive isolated West African hEbola virus described above, culturing the host cells to allow the virus to multiply, and harvesting the resulting virions. Also provided by the present invention are host cells infected with the inventive hEbola virus described above. In one embodiment of the present invention, the host cell is a primate cell.
[0070] In another aspect, the invention provides a method of detecting in a biological sample the presence of an antibody that immunospecifically binds hEbola virus, the method includes: (a) contacting the biological sample with the inventive host cell described above;
and (b) detecting the antibody bound to the cell.
[0071] In another aspect, the invention provides vaccine preparations, including the inventive hEbola virus, including recombinant and chimeric forms of the virus, nucleic acid molecules comprised by the virus, or protein subunits of the virus. In one embodiment, the vaccine preparations of the present invention includes live but attenuated hEbola virus with or without pharmaceutically acceptable carriers, including adjuvants. In another, the vaccine preparations of the invention comprise an inactivated or killed hEbola EboBun virus, EboIC
virus, or a combination thereof, with or without pharmaceutically acceptable carriers, including adjuvants. Such attenuated or inactivated viruses may be prepared by a series of passages of the virus through the host cells or by preparing recombinant or chimeric forms of virus. Accordingly, the present invention further provides methods of preparing recombinant or chimeric forms of the inventive hEbola viruses described herein.
[0072] In another specific embodiment, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of the inventive hEbola virus described above, and a pharmaceutically acceptable carrier. In another, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a protein extract of the inventive hEbola virus described above, or a subunit thereof;
and a pharmaceutically acceptable carrier. In another aspect, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof, and a pharmaceutically acceptable carrier. In another, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule comprising any of inventive the nucleotide sequences as described above, or a complement thereof, and a pharmaceutically acceptable carrier. In another aspect, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a protein extract of the inventive hEbola virus described above, or a subunit thereof; and a pharmaceutically acceptable carrier. In another aspect, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof, and a pharmaceutically acceptable carrier. In another, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule comprising any of inventive the nucleotide sequences as described above, or a complement thereof, and a pharmaceutically acceptable carrier.
[0073] In yet another specific embodiment, the vaccine preparations of the present invention comprise a nucleic acid or fragment of the hEbola virus, e.g., the virus having Accession No.
200706291, or nucleic acid molecules having the sequence of SEQ ID NOs: 1 or 10, or a fragment thereof. In another, the vaccine preparations comprise a polypeptide of the invention encoded by the nucleotide sequence of SEQ ID NOs: 1 or 10 or a fragment thereof. In a specific embodiment, the vaccine preparations comprise polypeptides of the invention as shown in SEQ ID
NOs: 2-9, 59, or 11-19, or encoded by the nucleotide sequence of SEQ ID NOs: 1 or 10, or a fragment thereof.
[0074] Furthermore, the present invention provides methods for treating, ameliorating, managing or preventing hemorrhagic fever by administering the vaccine preparations or antibodies of the present invention alone or in combination with adjuvants, or other pharmaceutically acceptable excipients. Furthermore, the present invention provides methods for treating, ameliorating, managing, or preventing hemorrhagic fever by administering the inventive compositions and formulations including the vaccine preparations or antibodies of the present invention alone or in combination with antivirals [e.g., amantadine, rimantadine, gancyclovir, acyclovir, ribavirin, penciclovir, oseltamivir, foscamet zidovudine (AZT), didanosine (ddl), lamivudine (3TC), zalcitabine (ddC), stavudine (d4T), nevirapine, delavirdine, indinavir, ritonavir, vidarabine, nelfinavir, saquinavir, relenza, tamiflu, pleconaril, interferons, etc.], steroids and corticosteroids such as prednisone, cortisone, fluticasone and glucocorticoid, antibiotics, analgesics, bronchodilators, or other treatments for respiratory and/or viral infections.
[0075] In a related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of the inventive hEbola virus described above, and a pharmaceutically acceptable carrier.
[0076] In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of a protein extract of the inventive hEbola virus described above or a subunit thereof, and a pharmaceutically acceptable carrier.
[0077] In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NOs: 1, 10, a combination thereof, or a complement thereof, and a pharmaceutically acceptable carrier.
[0078] In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of a nucleic acid molecule comprising the inventive nucleotide sequence as described above or a complement thereof, and a pharmaceutically acceptable carrier.
[0079] In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of any of the inventive polypeptides described above.
[0080] In another aspect, the present invention provides pharmaceutical compositions comprising antiviral agents of the present invention and a pharmaceutically acceptable carrier. In a specific embodiment, the antiviral agent of the invention is an antibody that immunospecifically binds hEbola virus or any hEbola epitope. In another specific embodiment, the antiviral agent is a polypeptide or protein of the present invention or nucleic acid molecule of the invention.
[0081] In a related aspect, the invention provides a pharmaceutical composition comprising a prophylactically or therapeutically effective amount of an anti-hEbola EboBun agent and a pharmaceutically acceptable carrier. In one embodiment of the present invention, the anti-hEbola EboBun agent is an antibody or an antigen-binding fragment thereof which immunospecifically binds to the hEbola virus of Deposit Accession No. 200706291, or polypeptides or protein derived therefrom. In another, the anti-hEbola agent is a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NOs: 1, 10, a combination thereof, or a fragment thereof.
In another, the anti-hEbola agent is a polypeptide encoded by a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NOs: 1, 10, a combination thereof, or a fragment thereof having a biological activity of the polypeptide.
[0082] The invention also provides kits containing compositions and formulations of the present invention. Thus, in another aspect, the invention provides a kit comprising a container containing the inventive immunogenic formulation described above.
[0083] In another aspect, the invention provides a kit includes a container containing the inventive vaccine formulation described above.
[0084] In another aspect, the invention provides a kit including a container containing the inventive pharmaceutical composition described above.
[0085] In another aspect, the invention provides a kit including a container containing the inventive vaccine formulation described above.
[0086] In another aspect, the invention provides a method for identifying a subject infected with the inventive hEbola virus described above, including: (a) obtaining total RNA
from a biological sample obtained from the subject; (b) reverse transcribing the total RNA to obtain cDNA; and (c) amplifying the cDNA using a set of primers derived from a nucleotide sequence of the inventive 5 hEbola virus described above.
[0087] In one embodiment of the present invention, the set of primers are derived from the nucleotide sequence of the genome of the hEbola virus of Deposit Accession No.
200706291. In another, the set of primers are derived from the nucleotide sequence of SEQ ID
NOs: 1 or 10 or any of the inventive nucleotide sequences as described above, or a complement thereof.
10 [0088] The invention further relates to the use of the sequence information of the isolated virus for diagnostic and therapeutic methods. In a specific embodiment, the invention provides nucleic acid molecules which are suitable for use as primers consisting of or including the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof, or at least a portion of the nucleotide sequence thereof. In another specific embodiment, the invention provides nucleic acid molecules 15 which are suitable for hybridization to the inventive hEbola nucleic acid;
including, but not limited to PCR primers, Reverse Transcriptase primers, probes for Southern analysis or other nucleic acid hybridization analysis for the detection of hEbola nucleic acids, e.g., consisting of or including the nucleotide sequence of SEQ ID NOs: 1, 10 a combination thereof, a complement thereof, or a portion thereof. The invention further encompasses chimeric or recombinant viruses encoded in 20 whole or in part by the nucleotide sequences.
[0089] In another aspect, the present invention provides methods for screening antiviral agents that inhibit the infectivity or replication of hEbola virus or variants thereof.
[0090] The invention further provides methods of preparing recombinant or chimeric forms of hEbola.
[0091] In another aspect, the invention provides vaccine preparations including the hEbola virus, including recombinant and chimeric forms of the virus, or subunits of the virus. The present invention encompasses recombinant or chimeric viruses encoded by viral vectors derived from the genome of the inventive hEbola virus described herein or natural variants thereof. In a specific embodiment, a recombinant virus is one derived from the hEbola virus of Deposit Accession No.
200706291. It is recognized that natural variants of the inventive hEbola viruses described herein comprise one or more mutations, including, but not limited to, point mutations, rearrangements, insertions, deletions etc., to the genomic sequence. It is recognized that the mutations may or may not result in a phenotypic change.
[0092] In another specific embodiment, a chimeric virus of the invention is a recombinant hEbola EboBun or EboIC virus which further comprises a heterologous nucleotide sequence. In accordance with the invention, a chimeric virus may be encoded by a nucleotide sequence in which heterologous nucleotide sequences have been added to the genome or in which endogenous or native nucleotide sequences have been replaced with heterologous nucleotide sequences.
[0093] According to the present invention, the chimeric viruses are encoded by the viral vectors of the invention which further comprise a heterologous nucleotide sequence. In accordance with the present invention a chimeric virus is encoded by a viral vector that may or may not include nucleic acids that are non-native to the viral genome. In accordance with the invention a chimeric virus is encoded by a viral vector to which heterologous nucleotide sequences have been added, inserted or substituted for native or non-native sequences. In accordance with the present invention, the chimeric virus may be encoded by nucleotide sequences derived from different species or variants of hEbola virus. In particular, the chimeric virus is encoded by nucleotide sequences that encode antigenic polypeptides derived from different species or variants of hEbola virus.
[0094] A chimeric virus may be of particular use for the generation of recombinant vaccines protecting against two or more viruses (Tao et al., J. Virol. 72, 2955-2961;
Durbin et al., 2000, J.
Virol. 74, 6821-6831; Skiadopoulos et al., 1998, J. Virol. 72, 1762-1768 (1998); Teng et al., 2000, J.
Virol. 74, 9317-9321). For example, it can be envisaged that a virus vector derived from the hEbola virus expressing one or more proteins of variants of hEbola virus including hEbola EboBun, or vice versa, will protect a subject vaccinated with such vector against infections by both the native hEbola and the variant. Attenuated and replication-defective viruses may be of use for vaccination purposes with live vaccines as has been suggested for other viruses. (See, for example, PCT WO 02/057302, at pp. 6 and 23; and United States Patent Application Publication 2008/0069838 incorporated by reference herein).
[0095] In accordance with the present invention the heterologous sequence to be incorporated into the viral vectors encoding the recombinant or chimeric viruses of the invention include sequences obtained or derived from different species or variants of hEbola.
[0096] In certain embodiments, the chimeric or recombinant viruses of the invention are encoded by viral vectors derived from viral genomes wherein one or more sequences, intergenic regions, termini sequences, or portions or entire ORF have been substituted with a heterologous or non-native sequence. In certain embodiments of the invention, the chimeric viruses of the invention are encoded by viral vectors derived from viral genomes wherein one or more heterologous sequences have been inserted or added to the vector.
[0097] The selection of the viral vector may depend on the species of the subject that is to be treated or protected from a viral infection. If the subject is human, then an attenuated hEbola virus can be used to provide the antigenic sequences.
[0098] In accordance with the present invention, the viral vectors can be engineered to provide antigenic sequences which confer protection against infection by the inventive hEbola and natural variants thereof. The viral vectors may be engineered to provide one, two, three or more antigenic sequences. In accordance with the present invention the antigenic sequences may be derived from the same virus, from different species or variants of the same type of virus, or from different viruses.
[0099] The expression products and/or recombinant or chimeric virions obtained in accordance with the invention may advantageously be utilized in vaccine formulations. The expression products and chimeric virions of the present invention may be engineered to create vaccines against a broad range of pathogens, including viral and bacterial antigens, tumor antigens, allergen antigens, and auto antigens involved in autoimmune disorders. One way to achieve this goal involves modifying existing hEbola genes to contain foreign sequences in their respective external domains. Where the heterologous sequences are epitopes or antigens of pathogens, these chimeric viruses may be used to induce a protective immune response against the disease agent from which these determinants are derived. In particular, the chimeric virions of the present invention may be engineered to create vaccines for the protection of a subject from infections with hEbola virus and variants thereof.
[00100] Thus, the present invention further relates to the use of viral vectors and recombinant or chimeric viruses to formulate vaccines against a broad range of viruses and/or antigens. The present invention also encompasses recombinant viruses including a viral vector derived from the hEbola or variants thereof which contains sequences which result in a virus having a phenotype more suitable for use in vaccine formulations, e.g., attenuated phenotype or enhanced antigenicity. The mutations and modifications can be in coding regions, in intergenic regions and in the leader and trailer sequences of the virus.
[00101] The invention provides a host cell including a nucleic acid or a vector according to the invention. Plasmid or viral vectors containing the polymerase components of hEbola virus are generated in prokaryotic cells for the expression of the components in relevant cell types (bacteria, insect cells, eukaryotic cells). Plasmid or viral vectors containing full-length or partial copies of the hEbola genome will be generated in prokaryotic cells for the expression of viral nucleic acids in vitro or in vivo. The latter vectors optionally contain other viral sequences for the generation of chimeric viruses or chimeric virus proteins, optionally lack parts of the viral genome for the generation of replication defective virus, and optionally contain mutations, deletions or insertions for the generation of attenuated viruses. In addition, the present invention provides a host cell infected with hEbola virus of Deposit Accession No. 200706291, [00102] Infectious copies of West African hEbola (being wild type, attenuated, replication-defective or chimeric) are optionally produced upon co-expression of the polymerase components according to the state-of-the-art technologies described above.
[0100] In addition, eukaryotic cells, transiently or stably expressing one or more full-length or partial hEbola proteins are optionally used. Such cells are preferably made by transfection (proteins or nucleic acid vectors), infection (viral vectors) or transduction (viral vectors) and are useful for complementation of mentioned wild type, attenuated, replication-defective or chimeric viruses.
[0101] The viral vectors and chimeric viruses of the present invention optionally modulate a subject's immune system by stimulating a humoral immune response, a cellular immune response or by stimulating tolerance to an antigen. As used herein, a subject means:
humans, primates, horses, cows, sheep, pigs, goats, dogs, cats, avian species and rodents.
Formulation of Vaccines and Antivirals [0102] In a preferred embodiment, the invention provides a proteinaceous molecule or hEbola virus specific viral protein or functional fragment thereof encoded by a nucleic acid according to the invention. Useful proteinaceous molecules are for example derived from any of the genes or genomic fragments derivable from the virus according to the invention, preferably the GP, L, NP, sGP, VP24, VP30, VP35, and VP 40 proteins described herein. Such molecules, or antigenic fragments thereof, as provided herein, are for example useful in diagnostic methods or kits and in pharmaceutical compositions such as subunit vaccines. Particularly useful are polypeptides encoded by the nucleotide sequence of SEQ ID NOs: 1 or 10; or antigenic fragments thereof for inclusion as antigen or subunit immunogen, but inactivated whole virus can also be used.
Particularly useful are also those proteinaceous substances that are encoded by recombinant nucleic acid fragments of the hEbola genome, of course preferred are those that are within the preferred bounds and metes of ORFs, in particular, for eliciting hEbola specific antibody or T cell responses, whether in vivo (e.g.
for protective or therapeutic purposes or for providing diagnostic antibodies) or in vitro (e.g. by phage display technology or another technique useful for generating synthetic antibodies).
[0103] It is recognized that numerous variants, analogues, or homologues of EboBun polypeptides are within the scope of the present invention including amino acid substitutions, alterations, modifications, or other amino acid changes that increase, decrease, or do not alter the function or immunogenic propensity of the inventive immunogen or vaccine.
Several post-translational modifications are similarly envisioned as within the scope of the present invention illustratively including incorporation of a non-naturally occurring amino acid(s), phosphorylation, glycosylation, sulfation, and addition of pendent groups such as biotynlation, fluorophores, lumiphores, radioactive groups, antigens, or other molecules.
[0104] Methods of expressing and purifying natural or recombinant peptides and proteins are well known in the art. Illustratively, peptides and proteins are recombinantly expressed in eukaryotic cells. Exemplary eukaryotic cells include yeast, HeLa cells, 293 cells, COS cells, Chinese hamster ovary cells (CHO), and many other cell types known in the art.
Both eukaryotic and prokaryotic expression systems and cells are available illustratively from Invitrogen Corp., Carlsbad, CA. It is appreciated that cell-free expression systems are similarly operable.
[0105] In a preferred embodiment an immunogenic polypeptide is a full length EboBun protein.
Preferably, an immunogen is a full length EboBun protein of SEQ ID NOs: 2-9 or 59, or EboIC SEQ
ID NOs: 11-19, or a fragment thereof as described herein. Preferably, an immunogen is has a minimum of 5 amino acids. As used herein an immunogen is preferably a polypeptide. In the context of an immunogenic polypeptide the terms immunogen, polypeptide, and antigen are used interchangeably.
[0106] Modifications and changes can be made in the structure of the inventive immunogens that are the subject of the application and still obtain a molecule having similar or improved characteristics as the wild-type sequence (e.g., a conservative amino acid substitution). For example, certain amino acids are optionally substituted for other amino acids in a sequence without appreciable loss of immunogenic activity. Because it is the interactive capacity and nature of a polypeptide that defines that polypeptide's biological functional activity, certain amino acid sequence substitutions can be made in a polypeptide sequence and nevertheless obtain a polypeptide with like or improved properties. Optionally, a polypeptide is used that has less or more immunogenic activity compared to the wild-type sequence.
[0107] In making such changes, the hydropathic index of amino acids is preferably considered.
The importance of the hydropathic amino acid index in conferring interactive biologic function on a polypeptide is generally understood in the art. It is known that certain amino acids can be substituted for other amino acids having a similar hydropathic index or score and still result in a 5 polypeptide with similar biological activity. Each amino acid has been assigned a hydropathic index on the basis of its hydrophobicity and charge characteristics. Those indices are: isoleucine (+4.5);
valine (+4.2); leucine (+3.8); phenylalanine (+2.8); cysteine/cysteine (+2.5);
methionine (+1.9);
alanine (+1.8); glycine (-0.4); threonine (-0.7); serine (-0.8); tryptophan (-0.9); tyrosine (-1.3);
proline (-1.6); histidine (-3.2); glutamate (-3.5); glutamine (-3.5);
aspartate (-3.5); asparagine (-3.5);
10 lysine (-3.9); and arginine (-4.5).
[0108] It is believed that the relative hydropathic character of the amino acid determines the secondary structure of the resultant polypeptide, which in turn defines the interaction of the polypeptide with other molecules, such as enzymes, substrates, receptors, antibodies, antigens, and the like. It is known in the art that an amino acid can be substituted by another amino acid having a 15 similar hydropathic index and still obtain a functionally equivalent immunogen. In such changes, the substitution of amino acids whose hydropathic indices are within 2 is preferred, those within 1 are particularly preferred, and those within 0.5 are even more particularly preferred.
[0109] As outlined above, amino acid substitutions are generally based on the relative similarity of the amino acid side-chain substituents, for example, their hydrophobicity, hydrophilicity, charge, 20 size, and the like. Exemplary substitutions that take various of the foregoing characteristics into consideration are well known to those of skill in the art and include (original residue: exemplary substitution): (Ala: Gly, Ser), (Arg: Lys), (Asn: Gln, His), (Asp: Glu, Cys, Ser), (Gln: Asn), (Glu:
Asp), (Gly: Ala), (His: Asn, Gln), (Ile: Leu, Val), (Leu: Ile, Val), (Lys:
Arg), (Met: Leu, Tyr), (Ser:
Thr), (Thr: Ser), (Tip: Tyr), (Tyr: Trp, Phe), and (Val: Ile, Leu).
Embodiments of this disclosure 25 thus contemplate functional or biological equivalents of a polypeptide and immunogen as set forth above. In particular, embodiments of the polypeptides and immunogens optionally include variants having about 50%, 60%, 70%, 80%, 90%, and 95% sequence identity to the polypeptide of interest.
[0110] The invention provides vaccine formulations for the prevention and treatment of infections with hEbola virus. In certain embodiments, the vaccine of the invention comprises recombinant and chimeric viruses of the hEbola virus. In certain embodiments, the virus is attenuated.
[0111] In another embodiment of this aspect of the invention, inactivated vaccine formulations are prepared using conventional techniques to "kill" the chimeric viruses.
Inactivated vaccines are "dead" in the sense that their infectivity has been destroyed. Ideally, the infectivity of the virus is destroyed without affecting its immunogenicity. In order to prepare inactivated vaccines, the chimeric virus may be grown in cell culture or in the allantois of the chick embryo, purified by zonal ultracentrifugation, inactivated by formaldehyde or (3-propiolactone, and pooled. The resulting vaccine is usually inoculated intramuscularly or intranasally.
[0112] Inactivated viruses are optionally formulated with a suitable adjuvant in order to enhance the immunological response. Such adjuvants illustratively include but are not limited to mineral gels, e.g., aluminum hydroxide; surface active substances such as lysolecithin, pluronic polyols, polyanions; peptides; oil emulsions; and potentially useful human adjuvants such as BCG and Corynebacterium parvum.
[0113] In another aspect, the present invention also provides DNA vaccine formulations including a nucleic acid or fragment of the inventive hEbola virus, e.g., the virus having Accession No. 200706291, or nucleic acid molecules having the sequence of SEQ ID NOs: 1 or 10, or a fragment thereof. In another specific embodiment, the DNA vaccine formulations of the present invention comprise a nucleic acid or fragment thereof encoding the antibodies which immunospecifically bind hEbola viruses. In DNA vaccine formulations, a vaccine DNA comprises a viral vector, such as that derived from the hEbola virus, bacterial plasmid, or other expression vector, bearing an insert including a nucleic acid molecule of the present invention operably linked to one or more control elements, thereby allowing expression of the vaccinating proteins encoded by the nucleic acid molecule in a vaccinated subject. Such vectors can be prepared by recombinant DNA technology as recombinant or chimeric viral vectors carrying a nucleic acid molecule of the present invention.
[0114] A nucleic acid as used herein refers to single- or double-stranded molecules which are optionally DNA, including the nucleotide bases A, T, C and G, or RNA, including the bases A, U
(substitutes for T), C, and G. The nucleic acid may represent a coding strand or its complement.
Nucleic acids are optionally identical in sequence to the sequence which is naturally occurring or include alternative codons which encode the same amino acid as that which is found in the naturally occurring sequence. Furthermore, nucleic acids optionally include codons which represent conservative substitutions of amino acids as are well known in the art.
[0115] As used herein, the term "isolated nucleic acid" means a nucleic acid separated or substantially free from at least some of the other components of the naturally occurring organism, for example, the cell structural components commonly found associated with nucleic acids in a cellular environment and/or other nucleic acids. The isolation of nucleic acids is illustratively accomplished by techniques such as cell lysis followed by phenol plus chloroform extraction, followed by ethanol precipitation of the nucleic acids. The nucleic acids of this invention are illustratively isolated from cells according to methods well known in the art for isolating nucleic acids. Alternatively, the nucleic acids of the present invention are optionally synthesized according to standard protocols well described in the literature for synthesizing nucleic acids. Modifications to the nucleic acids of the invention are also contemplated, provided that the essential structure and function of the peptide or polypeptide encoded by the nucleic acid are maintained.
[0116] The nucleic acid encoding the peptide or polypeptide of this invention is optionally part of a recombinant nucleic acid construct comprising any combination of restriction sites and/or functional elements as are well known in the art which facilitate molecular cloning and other recombinant DNA manipulations. Thus, the present invention further provides a recombinant nucleic acid construct including a nucleic acid encoding a polypeptide of this invention.
[0117] Generally, it may be more convenient to employ as the recombinant polynucleotide a cDNA version of the polynucleotide. It is believed that the use of a cDNA
version will provide advantages in that the size of the gene will generally be much smaller and more readily employed to transfect the targeted cell than will a genomic gene, which will typically be up to an order of magnitude larger than the cDNA gene. However, the inventor does not exclude the possibility of employing a genomic version of a particular gene where desired.
[0118] As used herein, the terms "engineered" and "recombinant" cells are synonymous with "host" cells and are intended to refer to a cell into which an exogenous DNA
segment or gene, such as a cDNA or gene has been introduced. Therefore, engineered cells are distinguishable from naturally occurring cells which do not contain a recombinantly introduced exogenous DNA segment or gene. A host cell is optionally a naturally occurring cell that is transformed with an exogenous DNA segment or gene or a cell that is not modified. A host cell preferably does not possess a naturally occurring gene encoding RSV G protein. Engineered cells are, thus, cells having a gene or genes introduced through the hand of man. Recombinant cells illustratively include those having an introduced cDNA or genomic DNA, and also include genes positioned adjacent to a promoter not naturally associated with the particular introduced gene.
[0119] To express a recombinant encoded polypeptide in accordance with the present invention one optionally prepares an expression vector that comprises a polynucleotide under the control of one or more promoters. To bring a coding sequence "under the control of' a promoter, one positions the 5' end of the translational initiation site of the reading frame generally between about 1 and 50 nucleotides "downstream" of (i.e., 3' of) the chosen promoter. The "upstream"
promoter stimulates transcription of the inserted DNA and promotes expression of the encoded recombinant protein.
This is the meaning of "recombinant expression" in the context used here.
[0120] Many standard techniques are available to construct expression vectors containing the appropriate nucleic acids and transcriptional/translational control sequences in order to achieve protein or peptide expression in a variety of host-expression systems. Cell types available for expression include, but are not limited to, bacteria, such as E. coli and B.
subtilis transformed with recombinant phage DNA, plasmid DNA or cosmid DNA expression vectors.
[0121] Certain examples of prokaryotic hosts illustratively include E. coli strain RR1, E. coli LE392, E. coli B, E. coli 1776 (ATCC No. 31537) as well as E. coli W3110 (F-, lambda-, prototrophic, ATCC No. 273325); bacilli such as Bacillus subtilis; and other enterobacteria such as Salmonella typhimurium, Serratia marcescens, and various Pseudomonas species.
[0122] In general, plasmid vectors containing replicon and control sequences that are derived from species compatible with the host cell are used in connection with these hosts. The vector ordinarily carries a replication site, as well as marking sequences that are capable of providing phenotypic selection in transformed cells. For example, E. coli is often transformed using pBR322, a plasmid derived from an E. coli species. Plasmid pBR322 contains genes for ampicillin and tetracycline resistance and thus provides easy means for identifying transformed cells. The pBR322 plasmid, or other microbial plasmid or phage may also contain, or be modified to contain, promoters that can be used by the microbial organism for expression of its own proteins.
[0123] In addition, phage vectors containing replicon and control sequences that are compatible with the host microorganism are optionally used as transforming vectors in connection with these hosts. For example, the phage lambda is optionally utilized in making a recombinant phage vector that can be used to transform host cells, such as E. coli LE392.
[0124] Further useful vectors include pIN vectors and pGEX vectors, for use in generating glutathione S-transferase (GST) soluble fusion proteins for later purification and separation or cleavage. Other suitable fusion proteins are those with (3-galactosidase, ubiquitin, or the like.
[0125] Promoters that are most commonly used in recombinant DNA construction include the (3-lactamase (penicillinase), lactose and tryptophan (trp) promoter systems.
While these are the most commonly used, other microbial promoters have been discovered and utilized, and details concerning their nucleotide sequences have been published, enabling those of skill in the art to ligate them functionally with plasmid vectors.
[0126] For expression in Saccharomyces, the plasmid YRp7, for example, is commonly used.
This plasmid contains the trpl gene, which provides a selection marker for a mutant strain of yeast lacking the ability to grow in tryptophan, for example ATCC No. 44076 or PEP4-1. The presence of the trpl lesion as a characteristic of the yeast host cell genome then provides an effective environment for detecting transformation by growth in the absence of tryptophan.
[0127] Suitable promoting sequences in yeast vectors illustratively include the promoters for 3-phosphoglycerate kinase or other glycolytic enzymes, such as enolase, glyceraldehyde-3-phosphate dehydrogenase, hexokinase, pyruvate decarboxylase, phosphofructokinase, glucose-6-phosphate isomerase, 3-phosphoglycerate mutase, pyruvate kinase, triosephosphate isomerase, phosphoglucose isomerase, and glucokinase. In constructing suitable expression plasmids, the termination sequences associated with these genes are also preferably ligated into the expression vector 3' of the sequence desired to be expressed to provide polyadenylation of the mRNA and termination.
[0128] Other suitable promoters, which have the additional advantage of transcription controlled by growth conditions, illustratively include the promoter region for alcohol dehydrogenase 2, isocytochrome C, acid phosphatase, degradative enzymes associated with nitrogen metabolism, and the aforementioned glyceraldehyde-3-phosphate dehydrogenase, and enzymes responsible for maltose and galactose utilization.
[0129] In addition to microorganisms, cultures of cells derived from multicellular organisms are also operable as hosts. In principle, any such cell culture is operable, whether from vertebrate or invertebrate culture. In addition to mammalian cells, these include insect cell systems infected with recombinant virus expression vectors (e.g., baculovirus); and plant cell systems infected with recombinant virus expression vectors (e.g., cauliflower mosaic virus, CaMV;
tobacco mosaic virus, TMV) or transformed with recombinant plasmid expression vectors (e.g., Ti plasmid) containing one or more coding sequences.
[0130] In a useful insect system, Autographica californica nuclear polyhedrosis virus (AcNPV) is used as a vector to express foreign genes. The virus grows in Spodoptera frugiperda cells. The isolated nucleic acid coding sequences are cloned into non-essential regions (for example the polyhedron gene) of the virus and placed under control of an AcNPV promoter (for example, the polyhedron promoter). Successful insertion of the coding sequences results in the inactivation of the polyhedron gene and production of non-occluded recombinant virus (i.e., virus lacking the proteinaceous coat coded for by the polyhedron gene). These recombinant viruses are then used to 5 infect Spodoptera frugiperda cells in which the inserted gene is expressed (e.g., U.S. Patent No.
4,215,051).
[0131] Examples of useful mammalian host cell lines include VERO and HeLa cells, Chinese hamster ovary (CHO) cell lines, W138, BHK, COS-7, 293, HepG2, NIH3T3, RIN and MDCK cell lines. In addition, a host cell is preferably chosen that modulates the expression of the inserted 10 sequences, or modifies and processes the gene product in the specific fashion desired. Such modifications (e.g., glycosylation) and processing (e.g., cleavage) of protein products may be important for the function of the encoded protein.
[0132] Different host cells have characteristic and specific mechanisms for the post-translational processing and modification of proteins. Appropriate cell lines or host systems are 15 preferably chosen to ensure the correct modification and processing of the foreign protein expressed.
Expression vectors for use in mammalian cells ordinarily include an origin of replication (as necessary), a promoter located in front of the gene to be expressed, along with any necessary ribosome binding sites, RNA splice sites, polyadenylation site, and transcriptional terminator sequences. The origin of replication is preferably provided either by construction of the vector to 20 include an exogenous origin, such as may be derived from SV40 or other viral (e.g., Polyoma, Adeno, VSV, BPV) source, or may be provided by the host cell chromosomal replication mechanism. If the vector is integrated into the host cell chromosome, the latter is often sufficient.
[0133] The promoters are optionally derived from the genome of mammalian cells (e.g., metallothionein promoter) or from mammalian viruses (e.g., the adenovirus late promoter; the 25 vaccinia virus 7.5K promoter). Further, it is also possible, and may be desirable, to utilize promoter or control sequences normally associated with the desired gene sequence, provided such control sequences are compatible with the host cell systems.
[0134] A number of viral based expression systems are operable herein, for example, commonly used promoters are derived from polyoma, Adenovirus 2, Adenovirus 5, cytomegalovirus and 30 Simian Virus 40 (SV40). The early and late promoters of SV40 virus are useful because both are obtained easily from the virus as a fragment which also contains the SV40 viral origin of replication.
Smaller or larger SV40 fragments are also operable, particularly when there is included the approximately 250 bp sequence extending from the HindIll site toward the Bgll site located in the viral origin of replication.
[0135] In cases where an adenovirus is used as an expression vector, the coding sequences are preferably ligated to an adenovirus transcription/translation control complex, e.g., the late promoter and tripartite leader sequence. This chimeric gene is then optionally inserted in the adenovirus genome by in vitro or in vivo recombination. Insertion in a non-essential region of the viral genome (e.g., region El or E3) will result in a recombinant virus that is viable and capable of expressing proteins in infected hosts.
[0136] Specific initiation signals may also be required for efficient translation of the claimed isolated nucleic acid coding sequences. These signals include the ATG
initiation codon and adjacent sequences. Exogenous translational control signals, including the ATG
initiation codon, may additionally need to be provided. One of ordinary skill in the art would readily be capable of determining this need and providing the necessary signals. It is well known that the initiation codon must be in-frame (or in-phase) with the reading frame of the desired coding sequence to ensure translation of the entire insert. These exogenous translational control signals and initiation codons are optionally of a variety of origins, both natural and synthetic. The efficiency of expression is optionally enhanced by the inclusion of appropriate transcription enhancer elements or transcription terminators.
[0137] In eukaryotic expression, one will also typically desire to incorporate into the transcriptional unit an appropriate polyadenylation site if one was not contained within the original cloned segment. Typically, the poly A addition site is placed about 30 to 2000 nucleotides "downstream" of the termination site of the protein at a position prior to transcription termination.
[0138] For long-term, high-yield production of recombinant proteins, stable expression is preferred. For example, cell lines that stably express constructs encoding proteins are engineered.
Rather than using expression vectors that contain viral origins of replication, host cells are preferably transformed with vectors controlled by appropriate expression control elements (e.g., promoter, enhancer, sequences, transcription terminators, polyadenylation sites, etc.), and a selectable marker.
Following the introduction of foreign DNA, engineered cells may be allowed to grow for 1-2 days in an enriched medium, and then are switched to a selective medium. The selectable marker in the recombinant plasmid confers resistance to the selection and allows cells to stably integrate the plasmid into their chromosomes and grow to form foci, which in turn can be cloned and expanded into cell lines.
[0139] A number of selection systems are illustratively used, including, but not limited, to the herpes simplex virus thymidine kinase, hypoxanthine-guanine phosphoribosyltransferase and adenine phosphoribosyltransferase genes, in tk-, hgprt- or aprt- cells, respectively. Also, antimetabolite resistance is optionally used as the basis of selection for dhfr, which confers resistance to methotrexate; gpt, which confers resistance to mycophenolic acid; neo, which confers resistance to the aminoglycoside G-418; and hygro, which confers resistance to hygromycin. It is appreciated that numerous other selection systems are known in the art that are similarly operable in the present invention.
[0140] The nucleic acids encoding the peptides and polypeptides of this invention are optionally administered as nucleic acid vaccines. For the purposes of vaccine delivery, a nucleic acid encoding a peptide or polypeptide of this invention is preferably in an expression vector that includes viral nucleic acid including, but not limited to, vaccinia virus, adenovirus, retrovirus and/or adeno-associated virus nucleic acid. The nucleic acid or vector of this invention is optoinally in a liposome or a delivery vehicle which can be taken up by a cell via receptor-mediated or other type of endocytosis. The nucleic acid vaccines of this invention are preferably in a pharmaceutically acceptable carrier or administered with an adjuvant. The nucleic acids encoding the peptides and polypeptides of this invention can also be administered to cells in vivo or ex vivo.
[0141] It is contemplated that the isolated nucleic acids of the disclosure are optionally "overexpressed", i.e., expressed in increased levels relative to its natural expression in cells of its indigenous organism, or even relative to the expression of other proteins in the recombinant host cell. Such overexpression is assessed by a variety of methods illustratively including radio-labeling and/or protein purification. However, simple and direct methods are preferred, for example, those involving SDS/PAGE and protein staining or immunoblotting, followed by quantitative analyses, such as densitometric scanning of the resultant gel or blot. A specific increase in the level of the recombinant protein or peptide in comparison to the level in natural in transfected cells is indicative of overexpression, as is a relative abundance of the specific protein in relation to the other proteins produced by the host cell and, e.g., visible on a gel.
[0142] Various heterologous vectors are described for DNA vaccinations against viral infections. For example, the vectors described in the following references, incorporated herein by reference, may be used to express hEbola sequences instead of the sequences of the viruses or other pathogens described; in particular, vectors described for hepatitis B virus (Michel, M. L. et al., 1995, DAN-mediated immunization to the hepatitis B surface antigen in mice: Aspects of the humoral response mimic hepatitis B viral infection in humans, Proc. Natl. Aca. Sci.
USA 92:5307-5311;
Davis, H. L. et al., 1993, DNA-based immunization induces continuous secretion of hepatitis B
surface antigen and high levels of circulating antibody, Human Molec. Genetics 2:1847-1851), HIV
virus (Wang, B. et al., 1993, Gene inoculation generates immune responses against human immunodeficiency virus type 1, Proc. Natl. Acad. Sci. USA 90:4156-4160; Lu, S.
et al., 1996, Simian immunodeficiency virus DNA vaccine trial in Macques, J. Virol. 70:3978-3991; Letvin, N.
L. et al., 1997, Potent, protective anti-HIV immune responses generated by bimodal HIV envelope DNA plus protein vaccination, Proc Natl Acad Sci USA. 94(17):9378-83), and influenza viruses (Robinson, H L et al., 1993, Protection against a lethal influenza virus challenge by immunization with a haemagglutinin-expressing plasmid DNA, Vaccine 11:957-960; Ulmer, J. B.
et al., Heterologous protection against influenza by injection of DNA encoding a viral protein, Science 259:1745-1749), as well as bacterial infections, such as tuberculosis (Tascon, R. E. et al., 1996, Vaccination against tuberculosis by DNA injection, Nature Med. 2:888-892;
Huygen, K. et al., 1996, Immunogenicity and protective efficacy of a tuberculosis DNA vaccine, Nature Med., 2:893-898), and parasitic infection, such as malaria (Sedegah, M., 1994, Protection against malaria by immunization with plasmid DNA encoding circumsporozoite protein, Proc. Natl.
Acad. Sci. USA
91:9866-9870; Doolan, D. L. et al., 1996, Circumventing genetic restriction of protection against malaria with multigene DNA immunization: CD8+T cell-interferon .delta., and nitric oxide-dependent immunity, J. Exper. Med., 1183:1739-1746).
[0143] Many methods are optionally used to introduce the vaccine formulations described above. These include, but are not limited to, oral, intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous, and intranasal routes. Alternatively, in a preferred embodiment the chimeric virus vaccine formulation is introduced via the natural route of infection of the pathogen for which the vaccine is designed. The DNA vaccines of the present invention are optionally administered in saline solutions by injections into muscle or skin using a syringe and needle (Wolff J. A. et al., 1990, Direct gene transfer into mouse muscle in vivo, Science 247:1465-1468; Raz, E., 1994, Intradermal gene immunization: The possible role of DNA uptake in the induction of cellular immunity to viruses, c. Natl. Acd. Sci. USA 91:9519-9523). Another way to administer DNA
vaccines operable herein is called the "gene gun" method, whereby microscopic gold beads coated with the DNA molecules of interest is fired into cells (Tang, D. et al., 1992, Genetic immunization is a simple method for eliciting an immune response, Nature 356:152-154). For general reviews of the methods for DNA vaccines, see Robinson, H. L., 1999, DNA vaccines: basic mechanism and immune responses (Review), Int. J. Mol. Med. 4(5):549-555; Barber, B., 1997, Introduction:
Emerging vaccine strategies, Seminars in Immunology 9(5):269-270; and Robinson, H. L. et al., 1997, DNA vaccines, Seminars in Immunology 9(5):271-283.
Attenuation of hEbola Virus or Variants Thereof [0144] The hEbola virus or variants thereof of the invention are optionally genetically engineered to exhibit an attenuated phenotype. In particular, the viruses of the invention exhibit an attenuated phenotype in a subject to which the virus is administered as a vaccine. Attenuation can be achieved by any method known to a skilled artisan. Without being bound by theory, the attenuated phenotype of the viruses of the invention is caused, e.g., by using a virus that naturally does not replicate well in an intended host species, for example, by reduced replication of the viral genome, by reduced ability of the virus to infect a host cell, or by reduced ability of the viral proteins to assemble to an infectious viral particle relative to the wild type species of the virus.
[0145] The attenuated phenotypes of hEbola virus or variants thereof are optionally tested by any method known to the artisan. A candidate virus, for example, is optionally tested for its ability to infect a host or for the rate of replication in a cell culture system. In certain embodiments, growth curves at different temperatures are used to test the attenuated phenotype of the virus. For example, an attenuated virus is able to grow at 35 C, but not at 39 C or 40 C. In certain embodiments, different cell lines are used to evaluate the attenuated phenotype of the virus. For example, an attenuated virus may only be able to grow in monkey cell lines but not the human cell lines, or the achievable virus titers in different cell lines are different for the attenuated virus. In certain embodiments, viral replication in the respiratory tract of a small animal model, including but not limited to, hamsters, cotton rats, mice and guinea pigs, is used to evaluate the attenuated phenotypes of the virus. In other embodiments, the immune response induced by the virus, including but not limited to, the antibody titers (e.g., assayed by plaque reduction neutralization assay or ELISA) is used to evaluate the attenuated phenotypes of the virus. In a specific embodiment, the plaque reduction neutralization assay or ELISA is carried out at a low dose. In certain embodiments, the ability of the hEbola virus to elicit pathological symptoms in an animal model is tested. A reduced ability of the virus to elicit pathological symptoms in an animal model system is indicative of its attenuated phenotype. In a specific embodiment, the candidate viruses are tested in a monkey model for nasal infection, indicated by mucus production.
[0146] The viruses of the invention are optionally attenuated such that one or more of the functional characteristics of the virus are impaired. In certain embodiments, attenuation is measured in comparison to the wild type species of the virus from which the attenuated virus is derived. In other embodiments, attenuation is determined by comparing the growth of an attenuated virus in 5 different host systems. Thus, for a non-limiting example, hEbola virus or a variant thereof is attenuated when grown in a human host if the growth of the hEbola or variant thereof in the human host is reduced compared to the non-attenuated hEbola or variant thereof.
[0147] In certain embodiments, the attenuated virus of the invention is capable of infecting a host, is capable of replicating in a host such that infectious viral particles are produced. In 10 comparison to the wild type species, however, the attenuated species grows to lower titers or grows more slowly. Any technique known to the skilled artisan can be used to determine the growth curve of the attenuated virus and compare it to the growth curve of the wild type virus.
[0148] In certain embodiments, the attenuated virus of the invention (e.g., a recombinant or chimeric hEbola) cannot replicate in human cells as well as the wild type virus (e.g., wild type 15 hEbola) does. However, the attenuated virus can replicate well in a cell line that lacks interferon functions, such as Vero cells.
[0149] In other embodiments, the attenuated virus of the invention is capable of infecting a host, of replicating in the host, and of causing proteins of the virus of the invention to be inserted into the cytoplasmic membrane, but the attenuated virus does not cause the host to produce new infectious 20 viral particles. In certain embodiments, the attenuated virus infects the host, replicates in the host, and causes viral proteins to be inserted in the cytoplasmic membrane of the host with the same efficiency as the wild type hEbola. In other embodiments, the ability of the attenuated virus to cause viral proteins to be inserted into the cytoplasmic membrane into the host cell is reduced compared to the wild type virus. In certain embodiments, the ability of the attenuated hEbola virus to replicate in 25 the host is reduced compared to the wild type virus. Any technique known to the skilled artisan can be used to determine whether a virus is capable of infecting a mammalian cell, of replicating within the host, and of causing viral proteins to be inserted into the cytoplasmic membrane of the host.
[0150] In certain embodiments, the attenuated virus of the invention is capable of infecting a host. In contrast to the wild type hEbola, however, the attenuated hEbola cannot be replicated in the 30 host. In a specific embodiment, the attenuated hEbola virus can infect a host and can cause the host to insert viral proteins in its cytoplasmic membranes, but the attenuated virus is incapable of being replicated in the host. Any method known to the skilled artisan can be used to test whether the attenuated hEbola has infected the host and has caused the host to insert viral proteins in its cytoplasmic membranes.
[0151] In certain embodiments, the ability of the attenuated virus to infect a host is reduced compared to the ability of the wild type virus to infect the same host. Any technique known to the skilled artisan can be used to determine whether a virus is capable of infecting a host.
[0152] In certain embodiments, mutations (e.g., missense mutations) are introduced into the genome of the virus, for example, into the sequence of SEQ ID NOs: 1 or 10, or to generate a virus with an attenuated phenotype. Mutations (e.g., missense mutations) can be introduced into the structural genes and/or regulatory genes of the hEbola. Mutations are optionally additions, substitutions, deletions, or combinations thereof. Such variant of hEbola can be screened for a predicted functionality, such as infectivity, replication ability, protein synthesis ability, assembling ability, as well as cytopathic effect in cell cultures. In a specific embodiment, the missense mutation is a cold-sensitive mutation. In another embodiment, the missense mutation is a heat-sensitive mutation. In another embodiment, the missense mutation prevents a normal processing or cleavage of the viral proteins.
[0153] In other embodiments, deletions are introduced into the genome of the hEbola virus, which result in the attenuation of the virus.
[0154] In certain embodiments, attenuation of the virus is achieved by replacing a gene of the wild type virus with a gene of a virus of a different species, of a different subgroup, or of a different variant. In another aspect, attenuation of the virus is achieved by replacing one or more specific domains of a protein of the wild type virus with domains derived from the corresponding protein of a virus of a different species. In certain other embodiments, attenuation of the virus is achieved by deleting one or more specific domains of a protein of the wild type virus.
[0155] When a live attenuated vaccine is used, its safety should also be considered. The vaccine preferably does not cause disease. Any techniques known in the art for improving vaccine safety are operable in the present invention. In addition to attenuation techniques, other techniques are optionally be used. One non-limiting example is to use a soluble heterologous gene that cannot be incorporated into the virion membrane. For example, a single copy of the soluble version of a viral transmembrane protein lacking the transmembrane and cytosolic domains thereof is used.
[0156] Various assays are optionally used to test the safety of a vaccine. For example, sucrose gradients and neutralization assays are used to test the safety. A sucrose gradient assay is optionally used to determine whether a heterologous protein is inserted in a virion. If the heterologous protein is inserted in the virion, the virion is preferably tested for its ability to cause symptoms in an appropriate animal model since the virus may have acquired new, possibly pathological, properties.
5.4 Adjuvants and Carrier Molecules [0157] hEbola-associated antigens are administered with one or more adjuvants.
In one embodiment, the hEbola-associated antigen is administered together with a mineral salt adjuvants or mineral salt gel adjuvant. Such mineral salt and mineral salt gel adjuvants include, but are not limited to, aluminum hydroxide (ALHYDROGEL, REHYDRAGEL), aluminum phosphate gel, aluminum hydroxyphosphate (ADJU-PHOS), and calcium phosphate.
[0158] In another embodiment, hEbola-associated antigen is administered with an immunostimulatory adjuvant. Such class of adjuvants include, but are not limited to, cytokines (e.g., interleukin-2, interleukin-7, interleukin-12, granulocyte-macrophage colony stimulating factor (GM-CSF), interferon-y interleukin-1(3 (IL-1 (3), and IL-1 0 peptide or Sclavo Peptide), cytokine-containing liposomes, triterpenoid glycosides or saponins (e.g., QuilA and QS-21, also sold under the trademark STIMULON, ISCOPREP), Muramyl Dipeptide (MDP) derivatives, such as N-acetyl-muramyl-L-threonyl-D-isoglutamine (Threonyl-MDP, sold under the trademark TERMURTIDE), GMDP, N-acetyl-nor-muramyl-L-alanyl-D-isoglutamine, N-acetylmuramyl-L-alanyl-D-isoglutaminyl-L-alanine-2-(1'-2'-dipalmitoyl-s- n-glycero-3-hydroxy phosphoryloxy)-ethylamine, muramyl tripeptide phosphatidylethanolamine (MTP-PE), unmethylated CpG
dinucleotides and oligonucleotides, such as bacterial DNA and fragments thereof, LPS, monophosphoryl Lipid A (3D-MLA sold under the trademark MPL), and polyphosphazenes.
[0159] In another embodiment, the adjuvant used is a particular adjuvant, including, but not limited to, emulsions, e.g., Freund's Complete Adjuvant, Freund's Incomplete Adjuvant, squalene or squalane oil-in-water adjuvant formulations, such as SAF and MF59, e.g., prepared with block-copolymers, such as L-121 (polyoxypropylene/polyoxyetheylene) sold under the trademark PLURONIC L-121, Liposomes, Virosomes, cochleates, and immune stimulating complex, which is sold under the trademark ISCOM.
[0160] In another embodiment, a microparticular adjuvant is used.
Microparticular adjuvants include, but are not limited to, biodegradable and biocompatible polyesters, homo- and copolymers of lactic acid (PLA) and glycolic acid (PGA), poly(lactide-co-glycolides) (PLGA) microparticles, polymers that self-associate into particulates (poloxamer particles), soluble polymers (polyphosphazenes), and virus-like particles (VLPs) such as recombinant protein particulates, e.g., hepatitis B surface antigen (HbsAg).
[0161] Yet another class of adjuvants that are optionally used include mucosal adjuvants, including but not limited to heat-labile enterotoxin from Escherichia coli (LT), cholera holotoxin (CT) and cholera Toxin B Subunit (CTB) from Vibrio cholerae, mutant toxins (e.g., LTK63 and LTR72), microparticles, and polymerized liposomes.
[0162] In other embodiments, any of the above classes of adjuvants are optionally used in combination with each other or with other adjuvants. For example, non-limiting examples of combination adjuvant preparations used to administer the hEbola-associated antigens of the invention include liposomes containing immunostimulatory protein, cytokines, T-cell and/or B-cell peptides, or microbes with or without entrapped IL-2 or microparticles containing enterotoxin.
Other adjuvants known in the art are also included within the scope of the invention (see Vaccine Design: The Subunit and Adjuvant Approach, Chap. 7, Michael F. Powell and Mark J. Newman (eds.), Plenum Press, New York, 1995, which is incorporated herein in its entirety).
[0163] The effectiveness of an adjuvant is illustratively determined by measuring the induction of antibodies directed against an immunogenic polypeptide containing a hEbola polypeptide epitope, the antibodies resulting from administration of this polypeptide in vaccines which are also comprised of the various adjuvants.
[0164] The polypeptides are optionally formulated into the vaccine as neutral or salt forms.
Pharmaceutically acceptable salts include the acid additional salts (formed with free amino groups of the peptide) and which are formed with inorganic acids, such as, for example, hydrochloric or phosphoric acids, or organic acids such as acetic, oxalic, tartaric, maleic, and the like. Salts formed with free carboxyl groups are optionally derived from inorganic bases, such as, for example, sodium potassium, ammonium, calcium, or ferric hydroxides, and such organic bases as isopropylamine, trimethylamine, 2-ethylamino ethanol, histidine, procaine and the like.
[0165] The vaccines of the invention are preferably multivalent or univalent.
Multivalent vaccines are made from recombinant viruses that direct the expression of more than one antigen.
[0166] Many methods are operable herein to introduce the vaccine formulations of the invention; these include but are not limited to oral, intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous, intranasal routes, and via scarification (scratching through the top layers of skin, e.g., using a bifurcated needle).
[0167] The patient to which the vaccine is administered is preferably a mammal, most preferably a human, but is also optionally a non-human animal including but not limited to lower primates, cows, horses, sheep, pigs, fowl (e.g., chickens), goats, cats, dogs, hamsters, mice and rats.
Preparation of Antibodies [0168] Antibodies that specifically recognize a polypeptide of the invention, such as, but not limited to, polypeptides including the sequence of SEQ ID NOs: 2-9, 59, or 11-19 and other polypeptides as described herein, or hEbola epitope or antigen-binding fragments thereof are used in a preferred embodiment for detecting, screening, and isolating the polypeptide of the invention or fragments thereof, or similar sequences that might encode similar enzymes from the other organisms. For example, in one specific embodiment, an antibody which immunospecifically binds hEbola epitope, or a fragment thereof, is used for various in vitro detection assays, including enzyme-linked immunosorbent assays (ELISA), radioimmunoassays, western blot, etc., for the detection of a polypeptide of the invention or, preferably, hEbola, in samples, for example, a biological material, including cells, cell culture media (e.g., bacterial cell culture media, mammalian cell culture media, insect cell culture media, yeast cell culture media, etc.), blood, plasma, serum, tissues, sputum, naseopharyngeal aspirates, etc.
[0169] Antibodies specific for a polypeptide of the invention or any epitope of hEbola are optionally generated by any suitable method known in the art. Polyclonal antibodies to an antigen of interest, for example, the hEbola virus from Deposit Accession No. 200706291, or including a nucleotide sequence of SEQ ID NOs: 1 or 10, are optionally produced by various procedures well known in the art. For example, an antigen is optionally administered to various host animals including, but not limited to, rabbits, mice, rats, etc., to induce the production of antisera containing polyclonal antibodies specific for the antigen. Various adjuvants are optionally used to increase the immunological response, depending on the host species, and include but are not limited to, Freund's (complete and incomplete) adjuvant, mineral gels such as aluminum hydroxide, surface active substances such as lysolecithin, pluronic polyols, polyanions, peptides, oil emulsions, keyhole limpet hemocyanins, dinitrophenol, and potentially useful adjuvants for humans such as BCG (Bacille Calmette-Guerin) and Corynebacterium parvum. Such adjuvants are also well known in the art.
[0170] Monoclonal antibodies are optionally prepared using a wide variety of techniques known in the art including the use of hybridoma, recombinant, and phage display technologies, or a combination thereof. In one example, monoclonal antibodies are produced using hybridoma techniques including those known in the art and taught, for example, in Harlow et al., Antibodies: A
Laboratory Manual (Cold Spring Harbor Laboratory Press, 2nd ed. 1988);
Hammerling, et al., in:
Monoclonal Antibodies and T-Cell Hybridomas, pp. 563-681 (Elsevier, N.Y., 1981) (both of which are incorporated by reference in their entireties). The term "monoclonal antibody" as used herein is not limited to antibodies produced through hybridoma technology. The term "monoclonal antibody"
refers to an antibody that is derived from a single clone, including any eukaryotic, prokaryotic, or phage clone, and not the method by which it is produced.
5 [0171] Methods for producing and screening for specific antibodies using hybridoma technology are routine and well known in the art. In a non-limiting example, mice are immunized with an antigen of interest or a cell expressing such an antigen. Once an immune response is detected, e.g., antibodies specific for the antigen are detected in the mouse serum, the mouse spleen is harvested and splenocytes isolated. The splenocytes are then fused by well known techniques to 10 any suitable myeloma cells. Hybridomas are selected and cloned by limiting dilution. The hybridoma clones are then assayed by methods known in the art for cells that secrete antibodies capable of binding the antigen. Ascites fluid, which generally contains high levels of antibodies, is optionally generated by inoculating mice intraperitoneally with positive hybridoma clones.
[0172] Antibody fragments which recognize specific epitopes are optionally generated by 15 known techniques. For example, Fab and F(ab')2 fragments are illustratively produced by proteolytic cleavage of immunoglobulin molecules, using enzymes such as papain (to produce Fab fragments) or pepsin (to produce F(ab')2 fragments). F(ab')2 fragments preferably contain the complete light chain, and the variable region, the CH1 region and the hinge region of the heavy chain.
[0173] The antibodies of the invention or fragments thereof are optionally produced by any 20 method known in the art for the synthesis of antibodies, in particular, by chemical synthesis or preferably, by recombinant expression techniques.
[0174] The nucleotide sequence encoding an antibody is obtained from any information available to those skilled in the art (i.e., from Genbank, the literature, or by routine cloning and sequence analysis). If a clone containing a nucleic acid encoding a particular antibody or an epitope-25 binding fragment thereof is not available, but the sequence of the antibody molecule or epitope-binding fragment thereof is known, a nucleic acid encoding the immunoglobulin may be chemically synthesized or obtained from a suitable source (e.g., an antibody cDNA
library, or a cDNA library generated from, or nucleic acid, preferably poly A+RNA, isolated from any tissue or cells expressing the antibody, such as hybridoma cells selected to express an antibody) by PCR
amplification using 30 synthetic primers hybridizable to the 3' and 5' ends of the sequence or by cloning using an oligonucleotide probe specific for the particular gene sequence to identify, e.g., a cDNA clone from a cDNA library that encodes the antibody. Amplified nucleic acids generated by PCR are optionally then cloned into replicable cloning vectors using any method known in the art.
[0175] Once the nucleotide sequence of the antibody is determined, the nucleotide sequence of the antibody is optionally manipulated using methods well known in the art for the manipulation of nucleotide sequences, e.g., recombinant DNA techniques, site directed mutagenesis, PCR, etc. (see, for example, the techniques described in Sambrook et al., supra;, and Ausubel et al., eds., 1998, Current Protocols in Molecular Biology, John Wiley & Sons, NY, which are both incorporated by reference herein in their entireties), to generate antibodies having a different amino acid sequence by, for example, introducing amino acid substitutions, deletions, and/or insertions into the epitope-binding domain regions of the antibodies or any portion of antibodies which may enhance or reduce biological activities of the antibodies.
[0176] Recombinant expression of an antibody requires construction of an expression vector containing a nucleotide sequence that encodes the antibody. Once a nucleotide sequence encoding an antibody molecule or a heavy or light chain of an antibody, or portion thereof has been obtained, the vector for the production of the antibody molecule is optionally produced by recombinant DNA
technology using techniques known in the art as discussed in the previous sections. Methods which are known to those skilled in the art are optionally used to construct expression vectors containing antibody coding sequences and appropriate transcriptional and translational control signals. These methods include, for example, in vitro recombinant DNA techniques, synthetic techniques, and in vivo genetic recombination. The nucleotide sequence encoding the heavy-chain variable region, light-chain variable region, both the heavy-chain and light-chain variable regions, an epitope-binding fragment of the heavy- and/or light-chain variable region, or one or more complementarity determining regions (CDRs) of an antibody are optionally cloned into such a vector for expression.
Thus, prepared expression vector is optionally then introduced into appropriate host cells for the expression of the antibody. Accordingly, the invention includes host cells containing a polynucleotide encoding an antibody specific for the polypeptides of the invention or fragments thereof.
[0177] The host cell is optionally co-transfected with two expression vectors of the invention, the first vector encoding a heavy chain derived polypeptide and the second vector encoding a light chain derived polypeptide. The two vectors illustratively contain identical selectable markers which enable equal expression of heavy and light chain polypeptides or different selectable markers to ensure maintenance of both plasmids. Alternatively, a single vector is optionally used which encodes, and is capable of expressing, both heavy and light chain polypeptides. In such situations, the light chain should be placed before the heavy chain to avoid an excess of toxic free heavy chain (Proudfoot, Nature, 322:52, 1986; and Kohler, Proc. Natl. Acad. Sci. USA, 77:2 197, 1980). The coding sequences for the heavy and light chains optionally include cDNA or genomic DNA.
[0178] In another embodiment, antibodies are generated using various phage display methods known in the art. In phage display methods, functional antibody domains are displayed on the surface of phage particles which carry the polynucleotide sequences encoding them. In a particular embodiment, such phage is utilized to display antigen binding domains, such as Fab and Fv or disulfide-bond stabilized Fv, expressed from a repertoire or combinatorial antibody library (e.g., human or murine). Phage expressing an antigen binding domain that binds the antigen of interest is optionally selected or identified with antigen, e.g., using labeled antigen or antigen bound or captured to a solid surface or bead. Phages used in these methods are typically filamentous phage, including fd and M13. The antigen binding domains are expressed as a recombinantly fused protein to either the phage gene III or gene VIII protein. Examples of phage display methods that can be used to make the immunoglobulins, or fragments thereof, of the present invention include those disclosed in Brinkman et al., J. Immunol. Methods, 182:41-50, 1995; Ames et al., J. Immunol.
Methods, 184:177-186, 1995; Kettleborough et al., Eur. J. Immunol., 24:952-958, 1994; Persic et al., Gene, 187:9-18, 1997; Burton et al., Advances in Immunology, 57:191-280, 1994;
PCT application No. PCT/GB91/01134; PCT publications WO 90/02809; WO 91/10737; WO 92/01047; WO
92/18619; WO 93/11236; WO 95/15982; WO 95/20401; and U.S. Pat. Nos. 5,698,426;
5,223,409;
5,403,484; 5,580,717; 5,427,908; 5,750,753; 5,821,047; 5,571,698; 5,427,908;
5,516,637;
5,780,225; 5,658,727; 5,733,743 and 5,969,108; each of which is incorporated herein by reference in its entirety.
[0179] As described in the above references, after phage selection, the antibody coding regions from the phage is optionally isolated and used to generate whole antibodies, including human antibodies, or any other desired fragments, and expressed in any desired host, including mammalian cells, insect cells, plant cells, yeast, and bacteria, e.g., as described in detail below. For example, techniques to recombinantly produce Fab, Fab' and F(ab')2 fragments are optionally employed using methods known in the art such as those disclosed in PCT publication WO
92/22324; Mullinax et al., BioTechniques, 12(6):864-869, 1992; and Sawai et al., AJRI, 34:26-34, 1995;
and Better et al., Science, 240:1041-1043, 1988 (each of which is incorporated by reference in its entirety). Examples of techniques operable to produce single-chain Fvs and antibodies include those described in U.S.
Pat. Nos. 4,946,778 and 5,258,498; Huston et al., Methods in Enzymology, 203:46-88, 1991; Shu et al., PNAS, 90:7995-7999, 1993; and Skerra et al., Science, 240:1038-1040, 1988.
[0180] Once an antibody molecule of the invention has been produced by any methods described above, or otherwise known in the art, it is then optionally purified by any method known in the art for purification of an immunoglobulin molecule, for example, by chromatography (e.g., ion exchange, affinity, particularly by affinity for the specific antigen after Protein A or Protein G
purification, and sizing column chromatography), centrifligation, differential solubility, or by any other standard technique(s) for the purification of proteins. Further, the antibodies of the present invention or fragments thereof are optionally fused to heterologous polypeptide sequences described herein or otherwise known in the art to facilitate purification. Illustrative examples include 6xHis tag, FLAG tag, biotin, avidin, or other system.
[0181] For some uses, including in vivo use of antibodies in humans and in vitro detection assays, it is preferable to use chimeric, humanized, or human antibodies. A
chimeric antibody is a molecule in which different portions of the antibody are derived from different animal species, such as antibodies having a variable region derived from a murine monoclonal antibody and a constant region derived from a human immunoglobulin. Methods for producing chimeric antibodies are known in the art. See e.g., Morrison, Science, 229:1202, 1985; Oi et al., BioTechniques, 4:214 1986; Gillies et al., J. Immunol. Methods, 125:191-202, 1989; U.S. Pat. Nos.
5,807,715; 4,816,567;
and 4,816,397, which are incorporated herein by reference in their entireties.
Humanized antibodies are antibody molecules from non-human species that bind the desired antigen having one or more complementarity determining regions (CDRs) from the non-human species and framework regions from a human immunoglobulin molecule. Often, framework residues in the human framework regions will be substituted with the corresponding residue from the CDR donor antibody to alter, preferably improve, antigen binding. These framework substitutions are identified by methods well known in the art, e.g., by modeling of the interactions of the CDR and framework residues to identify framework residues important for antigen binding and sequence comparison to identify unusual framework residues at particular positions. See, e.g., Queen et al., U.S. Pat. No. 5,585,089;
Riechmann et al., Nature, 332:323, 1988, which are incorporated herein by reference in their entireties. Antibodies are humanized using a variety of techniques known in the art including, for example, CDR-grafting (EP 239,400; PCT publication WO 91/09967; U.S. Pat. Nos.
5,225,539;
5,530,101 and 5,585,089), veneering or resurfacing (EP 592,106; EP 519,596;
Padlan, Molecular Immunology, 28(4/5):489-498, 1991; Studnicka et al., Protein Engineering, 7(6):805-814, 1994;
Roguska et al., Proc Natl. Acad. Sci. USA, 91:969-973, 1994), and chain shuffling (U.S. Pat. No.
5,565,332), all of which are hereby incorporated by reference in their entireties.
[0182] Completely human antibodies are particularly desirable for therapeutic treatment of human patients. Human antibodies are made by a variety of methods known in the art illustratively including phage display methods described above using antibody libraries derived from human immunoglobulin sequences. See U.S. Pat. Nos. 4,444,887 and 4,716,111; and PCT
publications WO
98/46645; WO 98/50433; WO 98/24893; WO 98/16654; WO 96/34096; WO 96/33735; and WO
91/10741, each of which is incorporated herein by reference in its entirety.
[0183] Human antibodies are also illustratively produced using transgenic mice which are incapable of expressing functional endogenous immunoglobulins, but which can express human immunoglobulin genes. For an overview of this technology for producing human antibodies, see Lonberg and Huszar, Int. Rev. Immunol., 13:65-93, 1995. For a detailed discussion of this technology for producing human antibodies and human monoclonal antibodies and protocols for producing such antibodies, see, e.g., PCT publications WO 98/24893; WO
92/01047; WO 96/34096;
WO 96/33735; European Patent No. 0 598 877; U.S. Pat. Nos. 5,413,923;
5,625,126; 5,633,425;
5,569,825; 5,661,016; 5,545,806; 5,814,318; 5,885,793; 5,916,771; and 5,939,598, which are incorporated by reference herein in their entireties. In addition, companies such as Abgenix, Inc.
(Fremont, Calif.), Medarex (NJ) and Genpharm (San Jose, Calif.) can be engaged to provide human antibodies directed against a selected antigen using technology similar to that described above.
[0184] Completely human antibodies which recognize a selected epitope are optionally generated using a technique referred to as "guided selection." In this approach a selected non-human monoclonal antibody, e.g., a mouse antibody, is used to guide the selection of a completely human antibody recognizing the same epitope. (Jespers et al., Bio/technology, 12:899-903, 1988).
[0185] Antibodies fused or conjugated to heterologous polypeptides are optionally used in in vitro immunoassays and in purification methods (e.g., affinity chromatography) known in the art.
See e.g., PCT publication No. WO 93/21232; EP 439,095; Naramura et al., Immunol. Lett., 39:91-99, 1994; U.S. Pat. No. 5,474,981; Gillies et al., PNAS, 89:1428-1432, 1992;
and Fell et al., J.
Immunol., 146:2446-2452, 1991, which are incorporated herein by reference in their entireties.
[0186] Antibodies may also be illustratively attached to solid supports, which are particularly useful for immunoassays or purification of the polypeptides of the invention or fragments, derivatives, analogs, or variants thereof, or similar molecules having the similar enzymatic activities as the polypeptide of the invention. Such solid supports include, but are not limited to, glass, cellulose, polyacrylamide, nylon, polystyrene, polyvinyl chloride or polypropylene.
Pharmaceutical Compositions and Kits 5 [0187] The present invention encompasses pharmaceutical compositions including antiviral agents of the present invention. In a specific embodiment, the antiviral agent is preferably an antibody which immunospecifically binds and neutralizes the hEbola virus or variants thereof, or any proteins derived therefrom. In another specific embodiment, the antiviral agent is a polypeptide or nucleic acid molecule of the invention. The pharmaceutical compositions have utility as an 10 antiviral prophylactic agent are illustratively administered to a subject where the subject has been exposed or is expected to be exposed to a virus.
[0188] Various delivery systems are known and operable to administer the pharmaceutical composition of the invention, illustratively, encapsulation in liposomes, microparticles, microcapsules, recombinant cells capable of expressing the mutant viruses, and receptor mediated 15 endocytosis (see, e.g., Wu and Wu, 1987, J. Biol. Chem. 262:4429 4432).
Methods of introduction include but are not limited to intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous, intranasal, epidural, and oral routes. The compounds may be administered by any convenient route, for example by infusion or bolus injection, by absorption through epithelial or mucocutaneous linings (e.g., oral mucosa, rectal and intestinal mucosa, etc.) and optionally administered together 20 with other biologically active agents. Administration is systemic or local.
In a preferred embodiment, it is desirable to introduce the pharmaceutical compositions of the invention into the lungs by any suitable route. Pulmonary administration can also be employed, e.g., by use of an inhaler or nebulizer, and formulation with an aerosolizing agent.
[0189] In a specific embodiment, it is desirable to administer the pharmaceutical compositions 25 of the invention locally to the area in need of treatment. This administration may be achieved by, for example, and not by way of limitation, local infusion during surgery, topical application, e.g., in conjunction with a wound dressing after surgery, by injection, by means of a catheter, by means of a suppository, by means of nasal spray, or by means of an implant, the implant being of a porous, non-porous, or gelatinous material, including membranes, such as sialastic membranes, or fibers. In one 30 embodiment, administration can be by direct injection at the site (or former site) infected tissues.
[0190] In another embodiment, the pharmaceutical composition is delivered in a vesicle, in particular a liposome (see Langer, 1990, Science 249:1527-1533; Treat et al., in Liposomes in the Therapy of Infectious Disease and Cancer, Lopez Berestein and Fidler (eds.), Liss, New York, pp.
353-365 (1989); Lopez-Berestein, ibid. , pp. 317-327; see generally ibid.).
[0191] In yet another embodiment, the pharmaceutical composition is delivered in a controlled release system. In one embodiment, a pump is used (see Langer, supra; Sefton, 1987, CRC Crit.
Ref. Biomed. Eng. 14:201; Buchwald et al., 1980, Surgery 88:507; and Saudek et al., 1989, N. Engl.
J. Med. 321:574). In another embodiment, polymeric materials are used (see Medical Applications of Controlled Release, Langer and Wise (eds.), CRC Pres., Boca Raton, Fla.
(1974); Controlled Drug Bioavailability, Drug Product Design and Performance, Smolen and Ball (eds.), Wiley, New York (1984); Ranger and Peppas, J. Macromol. Sci. Rev. Macromol. Chem. 23:61 (1983); see also Levy et al., 1985, Science 228:190; During et al., 1989, Ann. Neurol. 25:351;
Howard et al., 1989, J.
Neurosurg. 71:105). In yet another embodiment, a controlled release system is placed in proximity of the composition's target, i.e., the lung, thus, requiring only a fraction of the systemic dose (see, e.g., Goodson, in Medical Applications of Controlled Release, supra, vol. 2, pp. 115-138 (1984)).
[0192] Other controlled release systems are discussed in the review by Langer (Science 249:1527-1533 (1990)) the contents of which are incorporated herein by reference.
[0193] The pharmaceutical compositions of the present invention illustratively include a therapeutically effective amount of a live attenuated, inactivated or killed West African hEbola virus, or recombinant or chimeric hEbola virus, and a pharmaceutically acceptable carrier. In a specific embodiment, the term "pharmaceutically acceptable" means approved by a regulatory agency of the Federal or a state government or listed in the U.S. Pharmacopeia or other generally recognized pharmacopeia for use in animals, and more particularly in humans.
The term "carrier"
refers to a diluent, adjuvant, excipient, or vehicle with which the pharmaceutical composition is administered. Such pharmaceutical carriers are illustratively sterile liquids, such as water and oils, including those of petroleum, animal, vegetable or synthetic origin, such as peanut oil, soybean oil, mineral oil, sesame oil and the like. Water is a preferred carrier when the pharmaceutical composition is administered intravenously. Saline solutions and aqueous dextrose and glycerol solutions are optionally employed as liquid carriers, particularly for injectable solutions. Suitable pharmaceutical excipients include starch, glucose, lactose, sucrose, gelatin, malt, rice, flour, chalk, silica gel, sodium stearate, glycerol monostearate, talc, sodium chloride, dried skim milk, glycerol, propylene, glycol, water, ethanol and the like. The composition, if desired, also contains wetting or emulsifying agents, or pH buffering agents. These compositions optionally take the form of solutions, suspensions, emulsion, tablets, pills, capsules, powders, sustained release formulations and the like. The composition is optionally formulated as a suppository, with traditional binders and carriers such as triglycerides. Oral formulation illustratively includes standard carriers such as pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharine, cellulose, magnesium carbonate, etc. Examples of suitable pharmaceutical carriers are described in "Remington's Pharmaceutical Sciences" by E. W. Martin. The formulation should suit the mode of administration.
[0194] In a preferred embodiment, the composition is formulated in accordance with routine procedures as a pharmaceutical composition adapted for intravenous administration to human beings. Typically, compositions for intravenous administration are solutions in sterile isotonic aqueous buffer. The composition also includes an optional solubilizing agent and a local anesthetic such as lignocaine to ease pain at the site of the injection. Generally, the ingredients are supplied either separately or mixed together in unit dosage form, for example, as a dry lyophilized powder or water-free concentrate in a hermetically sealed container such as an ampoule or sachette indicating the quantity of active agent. Where the composition is to be administered by infusion, it can be dispensed with an infusion bottle containing sterile pharmaceutical grade water or saline. Where the composition is administered by injection, an ampoule of sterile water for injection or saline is optionally provided so that the ingredients may be mixed prior to administration.
[0195] The pharmaceutical compositions of the invention are illustratively formulated as neutral or salt forms. Pharmaceutically acceptable salts illustratively include those formed with free amino groups such as those derived from hydrochloric, phosphoric, acetic, oxalic, tartaric acids, etc., and those formed with free carboxyl groups such as those derived from sodium, potassium, ammonium, calcium, ferric hydroxides, isopropylamine, triethylamine, 2 ethylamino ethanol, histidine, procaine, etc.
[0196] The amount of the pharmaceutical composition of the invention which will be effective in the treatment of a particular disorder or condition will depend on the nature of the disorder or condition, and can be determined by standard clinical techniques. In addition, in vitro assays are optionally employed to help identify optimal dosage ranges. The precise dose to be employed in the formulation will also depend on the route of administration, and the seriousness of the disease or disorder, and should be decided according to the judgment of the practitioner and each patient's circumstances. However, suitable dosage ranges for intravenous administration are generally about 20 to 500 micrograms of active compound per kilogram body weight. Suitable dosage ranges for intranasal administration are generally about 0.01 pg/kg body weight to 1 mg/kg body weight.
Effective doses may be extrapolated from dose response curves derived from in vitro or animal model test systems.
[0197] Suppositories generally contain active ingredient in the range of 0.5%
to 10% by weight;
oral formulations preferably contain 10% to 95% active ingredient.
[0198] The invention also provides a pharmaceutical pack or kit including one or more containers filled with one or more of the ingredients of the pharmaceutical compositions of the invention. Optionally associated with such container(s) is a notice in the form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals or biological products, which notice reflects approval by the agency of manufacture, use or sale for human administration. In a preferred embodiment, the kit contains an antiviral agent of the invention, e.g., an antibody specific for the polypeptides encoded by a nucleotide sequence of SEQ ID NOs: 1 or 10, or as shown in SEQ ID NOs: 2-9, 59, or 11-19, or any hEbola epitope, or a polypeptide or protein of the present invention, or a nucleic acid molecule of the invention, alone or in combination with adjuvants, antivirals, antibiotics, analgesic, bronchodilators, or other pharmaceutically acceptable excipients.
[0199] The present invention further encompasses kits including a container containing a pharmaceutical composition of the present invention and instructions for use.
Detection Assays [0200] The present invention provides a method for detecting an antibody, which immunospecifically binds to the hEbola virus, in a biological sample, including for example blood, serum, plasma, saliva, urine, feces, etc., from a patient suffering from hEbola infection, and/or hemorrhagic fever. In a specific embodiment, the method including contacting the sample with the hEbola virus, for example, of Deposit Accession No. 200706291, or having a genomic nucleic acid sequence of SEQ ID NOs: 1 or 10, directly immobilized on a substrate and detecting the virus-bound antibody directly or indirectly by a labeled heterologous anti-isotype antibody. In another specific embodiment, the sample is contacted with a host cell which is infected by the hEbola virus, for example, of Deposit Accession No. 200706291, or having a genomic nucleic acid sequence of SEQ
ID NOs: 1 or 10, and the bound antibody is optionally detected by immunofluorescent assay.
[0201] An exemplary method for detecting the presence or absence of a polypeptide or nucleic acid of the invention in a biological sample involves obtaining a biological sample from various sources and contacting the sample with a compound or an agent capable of detecting an epitope or nucleic acid (e.g., mRNA, genomic DNA) of the hEbola virus such that the presence of the hEbola virus is detected in the sample. A preferred agent for detecting hEbola mRNA
or genomic RNA of the invention is a labeled nucleic acid probe capable of hybridizing to mRNA
or genomic RNA
encoding a polypeptide of the invention. The nucleic acid probe is, for example, a nucleic acid molecule including the nucleotide sequence of SEQ ID NOs: 1 or 10, a complement thereof, or a portion thereof, such as an oligonucleotide of at least 15, 20, 25, 30, 50, 100, 250, 500, 750, 1000 or more contiguous nucleotides in length and sufficient to specifically hybridize under stringent conditions to a hEbola mRNA or genomic RNA.
[0202] As used herein, the term "stringent conditions" describes conditions for hybridization and washing under which nucleotide sequences having at least 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, or 95% identity to each other typically remain hybridized to each other. Such hybridization conditions are described in, for example but not limited to, Current Protocols in Molecular Biology, John Wiley & Sons, N.Y. (1989), 6.3.1 6.3.6.;
Basic Methods in Molecular Biology, Elsevier Science Publishing Co., Inc., N.Y. (1986), pp.75 78, and 84 87; and Molecular Cloning, Cold Spring Harbor Laboratory, N.Y. (1982), pp.387 389, and are well known to those skilled in the art. A preferred, non-limiting example of stringent hybridization conditions is hybridization in 6x sodium chloride/sodium citrate (SSC), 0.5% SDS at about 68 C followed by one or more washes in 2xSSC, 0.5% SDS at room temperature. Another preferred, non-limiting example of stringent hybridization conditions is hybridization in 6x SSC at about 45 C
followed by one or more washes in 0.2x SSC, 0.1% SDS at 50 to 65 C.
[0203] A nucleic acid probe, polynucleotide, oligonucleotide, or other nucleic acid is preferably purified. An "isolated" or "purified" nucleotide sequence is substantially free of cellular material or other contaminating proteins from the cell or tissue source from which the nucleotide is derived, or is substantially free of chemical precursors or other chemicals when chemically synthesized. The language "substantially free of cellular material" includes preparations of a nucleotide/oligonucleotide in which the nucleotide/oligonucleotide is separated from cellular components of the cells from which it is isolated or produced. Thus, a nucleotide/oligonucleotide that is substantially free of cellular material includes preparations of the nucleotide having less than about 30%, 20%, 10%, 5%, 2.5%, or 1%, (by dry weight) of contaminating material. When nucleotide/oligonucleotide is produced by chemical synthesis, it is preferably substantially free of chemical precursors or other chemicals, i.e., it is separated from chemical precursors or other chemicals which are involved in the synthesis of the protein. Accordingly, such preparations of the nucleotide/oligonucleotide have less than about 30%, 20%, 10%, or 5% (by dry weight) of chemical precursors or compounds other than the nucleotide/oligonucleotide of interest.
In a preferred embodiment of the present invention, the nucleotide/oligonucleotide is isolated or purified.
[0204] In another preferred specific embodiment, the presence of hEbola virus is detected in the 5 sample by a reverse transcription polymerase chain reaction (RT-PCR) using the primers that are constructed based on a partial nucleotide sequence of the genome of hEbola virus, for example, that of Deposit Accession No. 200706291, or having a genomic nucleic acid sequence of SEQ ID NOs: 1 or 10. In a non-limiting specific embodiment, preferred primers to be used in a RT-PCR method are the primers are described in detail herein.
10 [0205] In more preferred specific embodiment, the present invention provides a real-time quantitative PCR assay to detect the presence of hEbola virus in a biological sample by subjecting the cDNA obtained by reverse transcription of the extracted total RNA from the sample to PCR
reactions using the specific primers described in detail herein, and a fluorescence dye, such as SYBR Green I, which fluoresces when bound nonspecifically to double-stranded DNA. The 15 fluorescence signals from these reactions are captured at the end of extension steps as PCR product is generated over a range of the thermal cycles, thereby allowing the quantitative determination of the viral load in the sample based on an amplification plot.
[0206] A preferred agent for detecting hEbola is an antibody that specifically binds a polypeptide of the invention or any hEbola epitope, preferably an antibody with a detectable label.
20 Antibodies are illustratively polyclonal, or more preferably, monoclonal.
An intact antibody, or a fragment thereof (e.g., Fab or F(ab')2) is operable herein.
[0207] The term "labeled", with regard to the probe or antibody, is intended to encompass direct labeling of the probe or antibody by coupling (i.e., physically linking) a detectable substance to the probe or antibody, optionally via a linker, as well as indirect labeling of the probe or antibody by 25 reactivity with another reagent that is directly labeled. Examples of indirect labeling include detection of a primary antibody using a fluorescently labeled secondary antibody and end-labeling of a DNA probe with biotin such that it is detectable with fluorescently labeled streptavidin. The detection method of the invention is optionally used to detect mRNA, protein (or any epitope), or genomic RNA in a sample in vitro as well as in vivo. Exemplary in vitro techniques for detection of 30 mRNA include northern hybridizations, in situ hybridizations, RT-PCR, and RNase protection. In vitro techniques for detection of an epitope of hEbola illustratively include enzyme linked immunosorbent assays (ELISAs), western blots, immunoprecipitations and immunofluorescence. In vitro techniques for detection of genomic RNA include northern hybridizations, RT-PCT, and RNase protection. Furthermore, in vivo techniques for detection of hEbola include introducing into a subject organism a labeled antibody directed against the polypeptide. In one embodiment, the antibody is labeled with a radioactive marker whose presence and location in the subject organism is detected by standard imaging techniques, including autoradiography.
[0208] In a specific embodiment, the methods further involve obtaining a control sample from a control subject, contacting the control sample with a compound or agent capable of detecting hEbola, e.g., a polypeptide of the invention or mRNA or genomic RNA encoding a polypeptide of the invention, such that the presence of hEbola or the polypeptide or mRNA or genomic RNA
encoding the polypeptide is detected in the sample, and comparing the absence of hEbola or the polypeptide or mRNA or genomic RNA encoding the polypeptide in the control sample with the presence of hEbola, or the polypeptide or mRNA or genomic DNA encoding the polypeptide in the test sample.
[0209] The invention also encompasses kits for detecting the presence of hEbola or a polypeptide or nucleic acid of the invention in a test sample. The kit illustratively includes a labeled compound or agent capable of detecting hEbola or the polypeptide or a nucleic acid molecule encoding the polypeptide in a test sample and, in certain embodiments, a means for determining the amount of the polypeptide or mRNA in the sample (e.g., an antibody which binds the polypeptide or an oligonucleotide probe which binds to DNA or mRNA encoding the polypeptide).
Kits optionally include instructions for use.
[0210] For antibody-based kits, the kit illustratively includes: (1) a first antibody (e.g., attached to a solid support) which binds to a polypeptide of the invention or hEbola epitope; and, optionally, (2) a second, different antibody which binds to either the polypeptide or the first antibody and is preferably conjugated to a detectable agent.
[0211] For oligonucleotide-based kits, the kit illustratively includes: (1) an oligonucleotide, e.g., a detectably labeled oligonucleotide, which hybridizes to a nucleic acid sequence encoding a polypeptide of the invention or to a sequence within the hEbola genome; or (2) a pair of primers useful for amplifying a nucleic acid molecule containing an hEbola sequence.
The kit optionally includes a buffering agent, a preservative, or a protein stabilizing agent.
The kit optionally includes components necessary for detecting the detectable agent (e.g., an enzyme or a substrate). The kit optionally contains a control sample or a series of control samples which can be assayed and compared to the test sample contained. Each component of the kit is usually enclosed within an individual container and all of the various containers are within a single package along with instructions for use.
Screening Assays to Identify Antiviral Agents [0212] The invention provides methods for the identification of a compound that inhibits the ability of hEbola virus to infect a host or a host cell. In certain embodiments, the invention provides methods for the identification of a compound that reduces the ability of hEbola virus to replicate in a host or a host cell. Any technique well known to the skilled artisan is illustratively used to screen for a compound useful to abolish or reduce the ability of hEbola virus to infect a host and/or to replicate in a host or a host cell.
[0213] In certain embodiments, the invention provides methods for the identification of a compound that inhibits the ability of hEbola virus to replicate in a mammal or a mammalian cell.
More specifically, the invention provides methods for the identification of a compound that inhibits the ability of hEbola virus to infect a mammal or a mammalian cell. In certain embodiments, the invention provides methods for the identification of a compound that inhibits the ability of hEbola virus to replicate in a mammalian cell. In a specific embodiment, the mammalian cell is a human cell.
[0214] In another embodiment, a cell is contacted with a test compound and infected with the hEbola virus. In certain embodiments, a control culture is infected with the hEbola virus in the absence of a test compound. The cell is optionally contacted with a test compound before, concurrently with, or subsequent to the infection with the hEbola virus. In a specific embodiment, the cell is a mammalian cell. In an even more specific embodiment, the cell is a human cell. In certain embodiments, the cell is incubated with the test compound for at least 1 minute, at least 5 minutes, at least 15 minutes, at least 30 minutes, at least 1 hour, at least 2 hours, at least 5 hours, at least 12 hours, or at least 1 day. The titer of the virus is optionally measured at any time during the assay. In certain embodiments, a time course of viral growth in the culture is determined. If the viral growth is inhibited or reduced in the presence of the test compound, the test compound is identified as being effective in inhibiting or reducing the growth or infection of the hEbola virus. In a specific embodiment, the compound that inhibits or reduces the growth of the hEbola virus is tested for its ability to inhibit or reduce the growth rate of other viruses to test its specificity for the hEbola virus.
[0215] In one embodiment, a test compound is administered to a model animal and the model animal is infected with the hEbola virus. In certain embodiments, a control model animal is infected with the hEbola virus without the administration of a test compound. The test compound is optionally administered before, concurrently with, or subsequent to the infection with the hEbola virus. In a specific embodiment, the model animal is a mammal. In an even more specific embodiment, the model animal is, but is not limited to, a cotton rat, a mouse, or a monkey. The titer of the virus in the model animal is optionally measured at any time during the assay. In certain embodiments, a time course of viral growth in the culture is determined. If the viral growth is inhibited or reduced in the presence of the test compound, the test compound is identified as being effective in inhibiting or reducing the growth or infection of the hEbola virus. In a specific embodiment, the compound that inhibits or reduces the growth of the hEbola in the model animal is tested for its ability to inhibit or reduce the growth rate of other viruses to test its specificity for the hEbola virus.
[0216] According to the method of the invention, a human or an animal is optionally treated for for EboBun or EboIC, other viral infection or bacterial infection by administering an effective amount of an inventive therapeutic composition. Preferably, a vaccine is administered prophylactically. An "effective amount" is an amount that will induce an immune response in a subject. Illustratively, an effective amount of the compositions of this invention ranges from nanogram/kg to milligram/kg amounts for young children and adults. Equivalent dosages for lighter or heavier body weights can readily be determined. The dose should be adjusted to suit the individual to whom the composition is administered and will vary with age, weight and metabolism of the individual. The exact amount of the composition required will vary from subject to subject, depending on the species, age, weight and general condition of the subject, the particular peptide or polypeptide used, its mode of administration and the like. An appropriate amount can be determined by one of ordinary skill in the art using only routine experimentation given the teachings herein. One skilled in the art will realize that dosages are best optimized by the practicing physician or veterinarian and methods for determining dose amounts and regimens and preparing dosage forms are described, for example, in Remington's Pharmaceutical Sciences, (Martin, E. W., ed., latest edition), Mack Publishing Co., Easton, PA. Preferably, a single administration is operable to induce an immune response.
[0217] Methods involving conventional biological techniques are described herein. Such techniques are generally known in the art and are described in detail in methodology treatises such as Molecular Cloning: A Laboratory Manual, 2nd ed., vol. 1-3, ed. Sambrook et al., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1989; and Current Protocols in Molecular Biology, ed. Ausubel et al., Greene Publishing and Wiley-Interscience, New York, 1992 (with periodic updates). Immunological methods (e.g., preparation of antigen-specific antibodies, immunoprecipitation, and immunoblotting) are described, e.g., in Current Protocols in Immunology, ed. Coligan et al., John Wiley & Sons, New York, 1991; and Methods of Immunological Analysis, ed. Masseyeff et al., John Wiley & Sons, New York, 1992.
[0218] Embodiments of inventive compositions and methods are illustrated in the following detailed examples. These examples are provided for illustrative purposes and are not considered limitations on the scope of inventive compositions and methods.
EXAMPLES
Example 1:
Newly discovered Ebola virus associated with hemorrhagic fever outbreak in Bundibugyo, Uganda [0219] In late November 2007 HF cases were reported in the townships of Bundibugyo and Kikyo in Bundibugyo District, Western Uganda (Figure 1A). These samples were assayed as described by Towner, JS, et al., PLoS Pathog, 2008 November; 4(11): e1000212, the contents of which are incorporated herein by reference for methods, results, reagents, and all other aspects of the publication. A total of 29 blood samples were initially collected from suspect cases and showed evidence of acute ebolavirus infection in eight specimens using a broadly reactive ebolavirus antigen capture assay known to cross-react with the different ebolavirus species and an IgM capture assay based on Zaire ebolavirus reagents (Table 1). These specimens were negative when initially tested with highly sensitive real-time RT-PCR assays specific for all known Zaire and Sudan ebolaviruses and marburgviruses. However, further evidence of acute ebolavirus infection was obtained using a traditionally less sensitive (relative to the real-time RT-PCR assays) but more broadly reactive filovirus L gene-specific RT-PCR assay (1 specimen) (Table 1). Sequence analysis of the PCR
fragment (400 bp of the virus L gene) revealed the reason for the initial failure of the real-time RT-PCR assays, as the sequence was distinct from that of the 4 known species of ebolavirus, although distantly related to Cote d'Ivoire ebolavirus. In total, 9 of 29 specimens showed evidence of ebolavirus infection, and all tests were negative for marburgvirus (data not shown).
[0220] Approximately 70% of the virus genome was rapidly sequenced from total RNA
extracted from a patient serum (#200706291) using a newly established metagenomics pyrosequencing method (454 Life Sciences) which involves successive rounds of random DNA
amplification8. Using the newly derived draft sequence, a real-time RT-PCR
assay specific for the 5 NP gene of this virus was quickly developed and evaluated. The assay was shown to have excellent sensitivity (Table 1), finding positive all the initial six samples that tested positive by either virus antigen capture (five specimens) or virus isolation assays (four specimens).
The antigen-capture, IgM, IgG and newly designed real-time PCR assays were quickly transferred to the Uganda Virus Research Institute during the course of the outbreak to facilitate rapid identification and isolation of 10 Ebola cases in the affected area for efficient control of the outbreak. The outbreak continued through late December 2007, and resulted in 149 suspected cases and 37 deaths9.
[0221] Table 1. Ebolavirus diagnostic results of initial 29 specimens obtained from Bundibugyo District with numerical specimen numbers assigned. RT-PCR refers to results obtained from conventional PCR using the broadly reactive Filo A/B primers 13. Ag, IgM, and IgG refer to 15 results from ELISA-based assaysio' 11 with Zaire ebolavirus reagents while virus isolation refers to culture attempts on Vero E6 cells12. Q-RT-PCR refers to results obtained using the optimized Bundibugyo ebolavirus specific real-time RT-PCR assay with cycle threshold (Ct) values of positive (Pos) samples indicated in the far right column. * Specimen #200706291 is the clinical sample from which prototype isolate #811250 was obtained.
Table 1 ...............................................................................
...............................................................................
..................................... .
Sample No. RT-PCR Arc Virus Isolation Q RT PCR Ct ......... .........
200706288 neg neg neg neg neg neg 40 200706289 neg neg neg neg neg neg 40 200706290....:...... n eg......>....neg.......n eg.......n eg.
_............neg ............................ n eg.....................
40.......
200706291.......... Pos......:.... Pos.......n eg neg Pos ............................ Pos..................23.64...>
200706292..........neg..........!?.eg.......!?...9.......11e9.
_............!?.eg ............. ..............neg..............
.......40.......
!?.eg .............. .............. neg..............
200706293........... eg...... ....!?.eg.......!?...9........ neg.
.............
.......40.......
200706294 neg neg neg neg neg neg 40 .................................
..................................................................
....................................... ....................................
.....................
200706295 neg neg neg neg neg neg 40 200706296 neg neg Pos Pos neg neg 40 200706297 neg neg Pos Pos neg neg 40 200706298 neg Pos Pos Pos neg Pos 34.83 200706299 neg neg Pos Pos neg neg 40 200706300............neg........... neg..... ne..... neg..
.............neg............................ neg.....................40 200706301 ne. neg ne neg neg ne. 40 .................................;..............................
.............;.......... ..................
................................... ...................................
200706302 neg Pos Pos neg neg Pos 35.01 ................................:.....................:........................
....................
........................................................................:......
...............
200706303 neg neg neg neg neg neg 40 200706304 neg neg neg neg Pos Pos 38.18 200706305....:...... neg......>....neg.......neg.......ne9 _............neg ............................ neg.....................40.......>
200706306..........neg......>....neg.......neg......ne9 _............neg ............................neg.....................40.......
200706307..........neg......>....!?eg.......neg......ne9_ _............neg ............................neg.....................40.......
200706320.... .......ND.......>....Pos.......neg.......neg. .............Pos Pos 30.24 ......................................... ..:..
200706321 ND neg neg neg neg neg 40 .................................
...................................................................
.......................................
..........................................................
200706322 ND neg neg neg neg neg 40 200706323 ND neg neg neg neg neg 40 200706324 ND neg neg neg neg neg 40 200706325 ND neg neg..... neg...... neg neg ..... 40 200706326 ND neg neg neg neg neg 40 200706327 ND Pos neg neg Pos Pos 34.41 200706328 ND neg...... neg......neg, neg ne9...... 40 [0222] The entire genome sequence of this virus was completed using a classic primer walking sequencing approach on RNA. The complete genome of the Eb ebolavirus was not available, so it too was derived by a similar combination of random primed pyrosequencing and primer walking approaches. Acquisition of these sequences allowed for the first time the phylogenetic analysis of the complete genomes of representatives of all known species of Ebola and Marburg viruses. The analysis revealed that the newly discovered virus differed from the four existing ebolavirus species (Figure 1), with approximately 32% nucleotide difference from even the closest relative, EboIC
(Table 2). Similar complete genome divergence (35-45%) is seen between the previously characterized ebolavirus species.
[0223] Table 2. Identity matrix based on comparisons of full-length genome sequences of Zaire ebolaviruses 1976 (Genbank accession number NC_002549) and 1995 (Genbank accession number AY354458), Sudan ebolavirus 2000 (Genbank accession number NC_006432), Cote d'Ivoire ebolavirus 1994 (SEQ ID NO: 10), Reston ebolavirus 1989 (Genbank accession number NC_004161), and Bundibugyo ebolavirus 2007 (SEQ ID NO: 1).
Table 2 Zaire `95 Sudan `00 EboIC `94 EboBun `07 Reston `89 Zaire `76 .988 .577 .630 .632 .581 Zaire `95 .577 .631 .633 .581 Sudan `00 .577 .577 .609 EboIC `94 .683 .575 EboBun `07 .576 [0224] The material and information obtained from the discovery of the new unique virus EboBun and the realization that together with EboIC these viruses represent a Glade of Bundibungyo-Ivory Coast Ebola virus species is valuable, and makes possible the development of clinical, diagnostic and research tools directed to human hEbola infection.
Material and Methods [0225] Ebolavirus detection and virus isolation. Several diagnostic techniques were used for each sample: (i) antigen capture, IgG, and IgM assays were performed as previously described" (ii) virus isolation attempts were performed on Vero E6 cells12 and monitored for 14 days; (iii) RNA
was extracted and tested for Zaire16 and Sudan ebolavirus and marburgvirus4 using real-time quantitative RT-PCR assays designed to detect all known species of each respective virus species the primers/probe for the Sudan ebolavirus assay were EboSudBMG 1(+) 5'-GCC ATG
GIT TCA GGT
TTG AG-3' (SEQ ID NO: 21), EboSudBMG 1(-) 5'-GGT IAC ATT GGG CAA CAA TTC A-3' (SEQ ID NO: 22) and Ebola Sudan BMG Probe 5'FAM-AC GGT GCA CAT TCT CCT TTT CTC
GGA-BHQ1 (SEQ ID NO: 23)]; (iv) the conventional RT-PCR was performed with the filo A/B
primer set as previously described16 using Superscript III (Invitrogen) according to the manufacturer's instructions. The specimen 200706291 was selected as the reference sample for further sequence analysis.
[0226] Genome sequencing. Pyrosequencing was carried out utilizing the approach developed by 454 Life Sciences, and the method described by Cox-Foster et al. 8 Subsequent virus whole genome primer walking was performed as previously described17 but using the primers specific for Bundibugyo ebolavirus RT-PCR amplification. In total, the entire virus genome was amplified in six overlapping RT-PCR fragments (all primers listed 5' to 3'): fragment A
(predicted size 2.7 kb) was amplified using forward-GTGAGACAAAGAATCATTCCTG (SEQ ID NO: 24) with reverse-CATCAATTGCTCAGAGATCCACC (SEQ ID NO: 25); fragment B (predicted size 3.0 kb) was amplified using forward-CCAACAACACTGCATGTAAGT (SEQ ID NO: 26) with reverse-AGGTCGCGTTAATCTTCATC (SEQ ID NO: 27); fragment C (predicted size 3.5 kb) was amplified using forward-GATGGTTGAGTTACTTTCCGG (SEQ ID NO: 28) with reverse-GTCTTGAGTCATCAATGCCC (SEQ ID NO: 29); fragment D (predicted size 3.1 kb) was amplified using forward-CCACCAGCACCAAAGGAC (SEQ ID NO: 30) with reverse-CTATCGGCAATGTAACTATTGG (SEQ ID NO: 31); fragment E (predicted size 3.4 kb) was amplified using forward-GCCGTTGTAGAGGACACAC (SEQ ID NO: 32) with reverse-CACATTAAATTGTTCTAACATGCAAG (SEQ ID NO: 33) and fragment F (predicted size 3.5 kb) was amplified using forward-CCTAGGTTATTTAGAAGGGACTA (SEQ ID NO: 34) with reverse-GGT AGA TGT ATT GAC AGC AAT ATC (SEQ ID NO: 35).
[0227] The exact 5' and 3' ends of Bundibugyo ebolavirus were determined by 3' RACE from virus RNA extracted from virus infected Vero E6 cell monolayers using TriPure isolation reagent.
RNAs were then polyadenylated in vitro using A-Plus poly(A) polymerase tailing kit (Epicenter Biotechnologies) following the manufacturer's instructions and then purified using an RNeasy kit (Qiagen) following standard protocols. Ten microliters of in vitro polyadenylated RNA were added as template in RT--PCR reactions, using SuperScript III One--Step R'I'-PCR
system with Platinum Tack High Fidelity (Invitrogen) following the manufacturer's protocol. Two parallel RT-PCR
reactions using the oligo(dT)-containing 3'RACf-AP primer (Invitrogen) mixed with I of 2 viral specific primers, Ebo-U 692(-) ACAAAAAGCTATCTGCACTAT (SEQ ID NO: 36) and Ebo-U18269(+) CTCAGAAGCAAAATTAATGG (SEQ ID NO: 37), generated -700 nt long fragments containing the 3' ends of either genomic and antigenomic RNAs. The resulting RT-PCR products were analyzed by agarose electrophoresis, and DNA bands of the correct sizes were purified using QlAquick Gel Extraction Kit (Qiagen) and sequenced using standard protocols (ABI).
[0228] The nucleotide sequence of the Cote d'Ivoire ebolavirus (EboIC) isolate RNA was initially determined using the exact same pyrosequencing strategy as that used for Bundibugyo ebolavirus described above. This method generated sequence for approximately 70% of the entire genome. This draft sequence was then used to design a whole genome primer walking strategy for filling any gaps and confirming the initial sequence. The following Cote d'Ivoire ebolavirus-specific primers were used to generate RT-PCR fragments, designated A-F, as follows: Fragment A
(predicted size 3.0 kb) was amplified using forward-GTGTGCGAATAACTATGAGGAAG
(SEQ
ID NO: 38) and reverse-GTCTGTGCAATGTTGATGAAGG (SEQ ID NO: 39); Fragment B
(predicted size 3.2 kb) was amplified using forward-CATGAAAACCACACTCAACAAC
(SEQ ID
NO: 40) and reverse-GTTGCCTTAATCTTCATCAAGTTC (SEQ ID NO: 41); Fragment C
(predicted size 3.0 kb) was amplified using forward-GGCTATAATGAATTTCCTCCAG
(SEQ ID
NO: 42) and reverse-CAAGTGTATTTGTGGTCCTAGC (SEQ ID NO: 43); fragment D
(predicted size 3.5 kb) was amplified using forward-GCTGGAATAGGAATCACAGG (SEQ ID NO: 44) and reverse-CGGTAGTCTACAGTTCTTTAG (SEQ ID NO: 45); fragment E (predicted size 4.0 kb) was amplified using forward-GACAAAGAGATTAGATTAGCTATAG (SEQ ID NO: 46) and reverse-GTAATGAGAAGGTGTCATTTGG (SEQ ID NO: 47); fragment F (predicted size 2.9 kb) was amplified using forward-CACGACTTAGTTGGACAATTGG (SEQ ID NO: 48) and reverse-CAGACACTAATTAGATCTGGAAG (SEQ ID NO: 49); fragment G (predicted size 1.3 kb) was amplified using forward-CGGACACACAAAAAGAAWRAA (SEQ ID NO: 50) and reverse-CGTTCTTGACCTTAGCAGTTC (SEQ ID NO: 51); and fragment H (predicted size 2.5 kb) was amplified using forward-GCACTATAAGCTCGATGAAGTC (SEQ ID NO: 52) and reverse-TGGACACACAAAAARGARAA (SEQ ID NO: 53). A gap in the sequence contig was located between fragments C and D and this was resolved using the following primers to generate a predicted fragment of 1.5 kb: forward-CTGAGAGGATCCAGAAGAAAG (SEQ ID NO: 54) and reverse-GTGTAAGCGTTGATATACCTCC (SEQ ID NO: 55). The terminal -20 nucleotides of the sequence were not experimentally determined but were inferred by comparing with the other known Ebola genome sequences.
[0229] Bundibugyo ebolavirus real-time RT-PCR assay. The primers and probe used in the Bundibugyo ebolavirus specific Q-RT-PCR assay were as follows: EboU965( +): 5'-GAGAAAAGGCCTGTCTGGAGAA-3' (SEQ ID NO: 56), EboU1039(-): 5'-TCGGGTATTGAATCAGACCTTGTT-3' (SEQ ID NO: 57) and EboU989 Prb: 5'Fam-TTCAACGACAAATCCAAGTGCACGCA-3'BHQ1 (SEQ ID NO 58). Q-RT-PCR reactions were set up using Superscript III One-Step Q-RT-PCR (Invitrogen) according to the manufacturer's instructions and run for 40 cycles with a 58 C annealing temperature.
[0230] Phylogenetic analysis. Modeltest 3.718 was used to examine 56 models of nucleotide substitution to determine the model most appropriate for the data. The General Time Reversible model incorporating invariant sites and a gamma distribution (GTR+I+G) was selected using the Akaike Information Criterion (AIC). Nucleotide frequencies were A = 0.3278, C
= 0.2101, G =
0.1832, T = 0.2789, the proportion of invariant sites = 0.1412, and the gamma shape parameter =
1.0593. A maximum likelihood analysis was subsequently performed in PAUP*4.ObIO19 using the GTR+I+G model parameters. Bootstrap support values were used to assess topological support and were calculated based on 1,000 pseudoreplicates20.
[0231] In addition, a Bayesian phylogenetic analysis was conducted in MrBayes 3.221 using the 5 GTR+I+G model of nucleotide substitution. Two simultaneous analyses, each with four Markov chains, were run for 5,000,000 generations sampling every 100 generations.
Prior to termination of the run, the AWTY module was used to assess Markov Chain Monte Carlo convergence to ensure that the length of the analysis was sufficient22. Trees generated before the stabilization of the likelihood scores were discarded (burn in = 40), and the remaining trees were used to construct a 10 consensus tree. Nodal support was assessed by posterior probability values (> 95 = statistical support).
Example 2 [0232] Immunization against EboBun:
15 [0233] To determine the capability of immunogens to elict an immune response in non-human primates (NHP), 12 cynomolgus macaques, of which 10 are immunized with VSVAG/EboBunGP
either orally (OR; n = 4), intranasally (IN; n = 4) or intramuscularly (IM; n = 2) in accordance with all animal control and safety guidelines and essentially as described by Qiu, X, et al., PLoS ONE. 2009;
4(5): e5547. The remaining 2 control animals are vaccinated intramuscularly with 20 VSVAG/MARVGP. VSVAG/MARVGP does not provide heterologous protection against EboBun, therefore these NHPs succumb to EboBun infection. Animals are acclimatized for 14 days prior to infection. Animals are fed and monitored twice daily (pre- and post-infection) and fed commercial monkey chow, treats and fruit. Husbandry enrichment consists of commercial toys and visual stimulation.
25 [0234] The recombinant VSVAG/EboBun vaccines are synthesized expressing the EboBun glycoprotein (GP) (SEQ ID NO: 9), soluble glycoprotein (sGP) (SEQ ID NO: 4), or nucleoprotein (NP) (SEQ ID NO: 3). Control VSVAG/MARVGP vaccines represent the analogous proteins from Lake victoria marburgvirus (MARV) (strain Musoke). The following results for GP are similar for sGP and NP. Vaccines are generated using VSV (Indiana serotype) as described previously.
30 Garbutt, M, et al., J Virol, 2004; 78(10):5458-5465; Schnell, MJ, et al., PNAS USA, 1996;
93(21):11359-11365. EboBun challenge virus is passaged in Vero E6 cells prior to challenge, as described previously Jones, SM, et al., Nat Med, 2005; 11(7):786-790;
Jahrling, PB, et al., J Infect Dis, 1999; 179(Suppl 1):S224-34. An EboBun immunogen peptide pool consisting of 15mers with 11 amino acid overlaps (Sigma-Genosys) spanning the entire sequence of the EboBun immunogens and strain Mayinga 1976 GP are used.
[0235] Twelve filovirus naive cynomolgus monkeys randomized into four groups receive 2 ml of lx107 PFU/ml of vaccine in Dulbecco's modified Eagle's medium (DMEM).
Animals in the three experimental groups are vaccinated with either: 1) 2 ml orally (OR) (n = 4);
2) 1 ml dripped into each nostril, intranasally (IN) (n = 4); or 3) 1 ml each into two sites intramuscularly (IM) (n = 2).
The two controls are injected intramuscularly with 2 ml of lx107 PFU/ml of VSVAG/MARVGP. All animals are challenged intramuscularly 28 days later with 1,000 PFU of EboBun.
[0236] Routine examination is conducted on 0, 2, 4, 6, 10, 14 and 21 days post-vaccination, then 0, 3, 6, 10, 14, 19, 26 days, 6 and 9 months after the EboBun challenge.
For the examinations animals are anaesthetized by intramuscular injection with 10 mg/kg of ketaset (Ayerst).
Examinations include haematological analysis, monitoring temperature (rectal), respiration rate, lymph nodes, weight, hydration, discharges and mucous membranes. Also, swabs (throat, oral, nasal, rectal, vaginal) and blood samples are collected (4 ml from femoral vein, 1 ml in EDTA vacutainer tube; 3 ml in serum separator vacutainer tube). Cynomolgus monkey PBMCs are isolated using BD
CPT sodium citrate Vacutainers (Becton Dickinson) as per manufacturer's protocol.
[0237] All VSVAG/EboBunGP immunized animals are protected from high dose challenge.
These animals show no evidence of clinical illness after vaccination or EboBun challenge. Both control animals demonstrate typical symptoms associated with EboBun HF
including fever, macular rashes, lethargy, and unresponsiveness. Continued infection requires euthanization. Hematology analyses at each examination date demonstrate increases in the platelet-crit in the OR and IN groups post-challenge, however, no significant changes are observed in any NHPs post-immunization or in the VSVAG/EboBunGP immunized NHPs post-challenge.
[0238] EboBun antibody production from humoral antibody response to vaccination and challenge is examined by a virus like particle (VLP) based ELISA assay.
Generation of EboBun VLPs is performed by the protocol for ZEBOV as described by Wahl-Jensen, V., et al., J Virol, 2005; 79(4):2413-2419. ELISA is performed by the protocol described by Qiu, X, et al., PLoS ONE.
2009; 4(5): e5547.
[0239] The VSVAG/MARVGP immunized animals do not develop a detectable antibody response to EboBun. In contrast, potent antibody responses are detected in all VSVAG/EboBunGP
immunized animals independent of immunization route. Between days 14 and 21 post-vaccination, all VSVAG/EboBunGP immunized NHPs develop high levels of IgA, IgM, and IgG
against EboBunGP. After challenge the IgM titres do not exceed the post-vaccination levels, however, IgG
and IgA antibody titres are increased peaking 14 days post-challenge then slowly decreasing before maintaining a relatively high antibody titre up to 9 months.
[0240] The level of neutralization antibodies is detected by a EboBun-GFP flow cytometric neutralization assay in serum collected at days 0 and 21 post-vaccination.
Samples are assayed in duplicate for their ability to neutralize an infection with EboBun-GFP in VeroE6 cells. Serially diluted serum samples are incubated with an equal volume of EboBun-GFP in DMEM, at 37 C, 5%
CO2 for 1 hr followed by addition of 150 l per well of a confluent 12 well plate of VeroE6 cells (MOI = 0.0005). After 2 hours at 37 C, 5% C02, 1 ml of DMEM, 2% fetal bovine serym (FBS), 100 U/ml penicillin, 100 g/ml streptomycin is added per well and incubated for 5 days. Cells are harvested by removing the culture supernatant, washing with 1 ml PBS, 0.04%
EDTA, then adding 800 l of PBS 0.04% EDTA for 5 minutes at 37 C before adding 8 ml PBS, 4%
paraformaldehyde (PFA) and overnight incubation. The cells are acquired (10,000 events) and analyzed with CellQuest Pro v3.3 on a Becton Dickinson FACSCalibur flow cytometer.
[0241] The OR and IN routes produce EboBunGP-specific neutralizing antibodies with the OR
route producing the highest titres post-vaccination. The IM immunization produces detectable levels of neutralizing antibody. In comparison, 3/4 NHPs in the OR group demonstrate a 50% reduction in EboBun-GFP positive cells at a titre of 1:40. Similarly, the IN route results in a reduction of EboBun-GFP positive cells at the 1:40 dilution.
[0242] EboBunGP-specific effector cellular immune responses are determined using IL-2 and IFN-y ELISPOT assays as described by Qin, X, et al., PLoS ONE. 2009; 4(5):
e5547 to determine the number of IL-2 and IFN-y secreting lymphocytes. Prior to challenge on days 10 to 14 post-vaccination there is a detectable EboBun immunogen-specific IFN-y response in all immunized animals. The IM route is the most potent, inducing approximately 2-fold more IFN-y secreting cells than OR (p<0.001) or IN (p = 0.043) routes. A strong post-challenge secondary IFN-y response is induced in all VSVAG/EboBun immunized animals with the IM route producing the most IFN-y cells at day 6. By day 10 the OR group demonstrates a stronger response. The IFN-y in the IN
group rises steadily, peaking at day 26 post-challenge with 4.3 and 2 fold more EboBun specific IFN-y secreting cells than the IM (p = 0.003) and OR (p = 0.075) group, respectively. All three routes produce strong EboBun-specific IFN-y responses.
[0243] Post-vaccination, the IM group also has more EboBunGP-specific IL-2 secreting cells than either of the mucosally immunized groups. Post-challenge, the IM route continues to dominate early after challenge peaking on day 10. This difference shows a trend when compared to the IN
group (p = 0.067) and is significant when compared to the OR group (p<0.001).
Additionally, the IN
group has more IL-2 producing cells than the OR group (p = 0.090) on day 10 post-challenge. By day 26 post-challenge all three routes continue to produce a EboBunGP-specific IL-2 response, however, the IN group response is strongest. At day 26 post-challenge the IN
group has the most potent IFN-y and IL-2 responses, as well as the highest IgA and IgG antibody titre, indicating this immunization route, followed by a EboBun challenge, results in the development of potent and sustained effector responses.
[0244] Absolute lymphocyte numbers for CD3+, CD4+, and CD8+ (CD3+4-) T cell populations are determined by flow cytometry. No decrease is observed in the lymphocyte populations for any of the VSVAG/EboBunGP vaccinated NHPs. In contrast, control animals who are not protected from EboBun show lymphocyte numbers decreased by 28-57%.
[0245] Macrophage numbers are slightly increased in control animals. However, the number of CD14+ cells is greater in the VSVAG/EboBunGP vaccinated groups with the IM
route showing the most significant increases.
[0246] In order to determine the long term immune response after challenge, EboBunGP-specific CD4+ and CD8+ memory T-lymphocytes are examined for their ability to proliferate (CFSE-) or produce IFN-y in response to EboBunGP peptides at 6 months post-vaccination.
EboBunGP-specific memory responses are observed as a result of vaccination followed by a ZEBOV challenge. These responses persist for at least 6 months. The memory populations in OR
and IN inoculation routes demonstrate the greatest potential for proliferation and IFN-y production post-challenge.
[0247] Any patents or publications mentioned in this specification are incorporated herein by reference to the same extent as if each individual publication is specifically and individually indicated to be incorporated by reference.
[0248] The compositions and methods described herein are presently representative of preferred embodiments, exemplary, and not intended as limitations on the scope of the invention. Changes therein and other uses will occur to those skilled in the art. Such changes and other uses can be made without departing from the scope of the invention as set forth in the claims. All numerical ranges are inclusive of the whole integers and decimals between the endpoints, and inclusive of the endpoints.
References 1. Suzuki, Y., and Gojobori, T., (1997) The origin and evolution of Ebola and Marburg viruses. Mol Bio Evol, 14(8): 800-806.
2. Sanchez, A., Geisbert, T.W., Feldmann, H. in Fields Virology (ed. Knipe, D.
M., Howley, P.M.) 1409 - 1448 (Lippincott Williams and Wilkins, Philadelphia, 2007).
310 Leroy, E.M. et al., (2005) Fruit bats as reservoirs of Ebola virus.
Nature, 438, 575-6.
4. Towner, J.S. et al., (2007) Marburg virus infection detected in a common African bat. PLoS ONE, 2(8), e764.
5. Swanepoel, R. et al., (2007) Studies of reservoir hosts for Marburg virus.
Emerg Infect Dis, 13(12), 1847-51.
615 Le Guenno, B. et al., (1995) Isolation and partial characterization of a new species of Ebola virus.
Lancet, 345(8960), 1271-4.
7. Ksiazek, T. G. et al. (1999) Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, IgG and IgM antibody findings among EHF patients in Kikwit, 1995. J.
Infect Dis 179 (suppl 1), S177-S187.
820 Cox-Foster, D. L. et al. (2007) A metagenomic survey of microbes in honey bee colony collapse disorder. Science 318, 283-7.
9. World Health Organization (2008) Ebola outbreak contained in Uganda.
Features, 22 February, www.who.int/features/2008/ebola-outbreak/en/.
10. Sullivan, N. J., Sanchez, A., Rollin, P. E., Yang, Z.-Y. & Nabel, G. J.
(2000) Development of a 25 preventive vaccine for Ebola virus infection in primates. Nature 408, 605-609.
11. Ksiazek, T. G., West, C. P., Rollin, P. E., Jahrling, P. B. & Peters, C.
J. (1999) ELISA for the detection of antibodies to Ebola viruses. J. Infect Dis 179 (suppl 1), S191-S198.
12. Rodriguez, L. et al. (1999) Persistence and genetic stability of Ebola virus during the outbreak in Kikwit, Zaire 1995. J. Infect Dis 179 (suppl 1), S170-S176.
130 Sanchez, A. et al. Detection and molecular characterization of Ebola viruses causing disease in human and nonhuman primates. J. Infect Dis 179 (suppl 1), S164-S169 (1999).
14. Jones, S. M. et al. (2005) Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat Med 11, 786-90.
15. Geisbert, T. W. et al. (2008) Recombinant vesicular stomatitis virus vector mediates postexposure protection against Sudan Ebola hemorrhagic fever in nonhuman primates. J Virol 82, 5664-8.
165 Towner, J. S., Sealy, T. K., Ksiazek, T. & Nichol, S. T. (2007) High-throughput molecular detection of hemorrhagic fever virus threats with applications for outbreak settings. J.
Inf Dis 196 (suppl 2), S205-212.
17. Towner, J. S. et al. (2006) Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J Virol 80, 6497-516.
1$0 Posada, D. & Crandall, K. A. (1998) MODELTEST: testing the model of DNA
substitution.
Bioinformatics 14, 817-818.
19. Swofford, D. L. (2002) PAUP*: phylogenetic analysis using parsimony (*and other methods) version 4.0blO. Sinauer Assoc., Sunderland, Mass.
20. Felsenstein, J. (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 15 39, 783-791.
21. Ronquist, F. & Huelsenbeck, J. P. (2003) MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572-1574.
22. Nylander, J. A. A., Wilgenbusch, J. C., Warren, D. L. & Swofford, D. L.
(2008) AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics.
20 Bioinformatics 24, 581-583.
Tässä alla on USA:n hallituksen kehittämä hiv-viruksen patentti, jolla USA on käynyt sotaan ihmiskuntaa vastaan. USA:n hallitus on pyrkinyt tappamaan tällä viruksella afrikkalaiset ja myös muut.
http://www.patents.com/us-4647773.html


United States Patent
4,647,773 Gallo , et al.March 3, 1987
References Cited
U.S. Patent Documents
Other References
Primary Examiner: Warren; Charles F.
Assistant Examiner: Tarcza; John Edward
Attorney, Agent or Firm: Roberts, Jr.; John S.
Claims
We claim:
1. A method for continuous production of HTLV-III virus which comprises infecting in cell culture highly susceptible, permissive cells consisting of a neoplastic aneuploid HT-cell line with said virus, said cells preserve the capacity for permanent growth after the infection with said virus, growing said cells under conditions suitable for cell growth and recovering said virus produced by said cells.
2. The method of claim 1, wherein said virus consists of cytopathic variants of HTLV.
3. The method of claim 1 wherein said infecting comprises cocultivating said virus with said cells to produce a cell line and recovering said cell line.
4. The method of claim 1 wherein said infecting comprises cell-free infection of said cells with said virus.
5. The method of claim 1 wherein said cells are neoplastic aneuploid T-cells derived from an adult with lymphoid leukemia.
6. A process for the continuous production of HTLV-III virus which comprises cocultivating said virus with a target HT-cell to produce an infected cell line, growing said cell line and recovering said virus from supernatants produced by said cell line.
7. The process of claim 6 wherein said target T-cell is a neoplastic aneuploid T-cell susceptible to infection with HTLV-III.
8. A process for the continual production of HTLV-III by infecting T-cells in cultures which comprises cocultivating HTLV-III virus with an HT-clone to produce an infected cell line, said clone being an aneuploid T-cell line derived from lymphoid leukemia, growing said cell line and recovering said virus from supernatants produced by said cell line.
9. The process in claim 8 wherein said clone is a mature T-cell phenotype of OKT3.sup.+ (62%), OKT4.sup.+ (39%) and OKT8.sup.-.
10. A method of producing a cell line containing an antigen of HTLV-III which comprises infecting an HT-cell line derived from lymphoid leukemia and susceptible to infection with HTLV-III, said cell line is capable of continuous large-scale production of HTLV-III, and growing the infected cell line under conditions suitable for growth.
11. The method of claim 10 wherein said cell line is a neoplastic aneuploid T-cell line.
12. The method of claim 10 wherein said HTLV-III are variants of human T-lymphotropic retrovirus, exhibit cytopathic effects and are non-transforming.
13. A cell line containing HTLV-III designated H9/HTLV-III.sub.B, ATCC Accession CRL 8543.
14. A process for producing a cell line H9/HTLV-III.sub.B which comprises infecting a target T-cell with HTLV-III virus to produce a cell line and recovering same, said infecting process overcomes the normal cytopathic effect of HTLV-III and preserves the immortal growth capacity of the target cell.
15. An HT cell line permanently infected with HTLV-III virus, wherein said cell line continually produces said virus.
Description
The present invention describes a cell system for the reproducible detection and isolation of human T-lymphotropic retroviruses (HTLV-family) with cytopathic or cell killing effects (HTLV-III) from patients with the acquired immune deficiency syndrome (AIDS), pre-AIDS and in healthy carriers. One neoplastic aneuploid T-cell line derived from an adult with lymphoid leukemia, and termed HT, was susceptible to infection with the new variants of HTLV, providing T-cell populations which are highly susceptible and permissive for HTLV-III, and convenience for large scale production, isolation, and biological detection of the virus.
BACKGROUND OF THE INVENTION
The disclosure of this invention is contained in the following journal articles: Gallo et al., "Detection, Isolation, and Continuous Production of Cytopathic Human T-Lymphotropic Retroviruses (HTLV-III) from Patients with AIDS and pre-AIDS," Science, in press; and Gallo et al., "Human T-Lymphotropic Retrovirus, HTLV-III, Isolated from AIDS Patients and Donors at Risk for AIDS," in press.
Epidemiologic data strongly suggest that acquired immune deficiency syndrome (AIDS) is caused by an infectious agent which is apparently horizontally transmitted by intimate contact or blood products. Though the disease is manifested by opportunistic infections, predominantly Pneumocystis carcinii pneumonia and Kaposi's sarcoma, the underlying disorder affects the patient's cell-mediated immunity with absolute lymphopenia and reduced helper T-lymphocyte (OKT4.sup.+) subpopulation(s). Moreover, before a complete clinical manifestation of the disease occurs, its prodrome, pre-AIDS, is frequently characterized by unexplained chronical lymphadenopathy and/or leukopenia involving a helper T cell subset. This leads to the severe immune deficiency of the patient, suggesting that a specific subset of T-cells is the primary target for an infectious agent. Although patients with AIDS or pre-AIDS are often chronically infected with cytomegalovirus or hepatitis B virus, for various reasons these appear to be opportunistic or coincidental infections apparently not linked to the immunological response deficiency. It is believed that the cause of AIDS may be a virus from the family of human T-cell lymphotropic retroviruses (HTLV) which, prior to the present invention, comprised two major well characterized subgroups of human retroviruses, called human T-cell leukemia/lymphoma viruses, HTLV-I and HTLV-II. The most common isolate, HTLV-I, is mainly obtained from patients with mature T-cell malignancies. Seroepidemiological studies, in vitro biological effects, and nucleic acid hybridization data indicate that HTLV-I is etiologically associated with these malignancies, affecting adults primarily in the south of Japan, the Caribbean and Africa. HTLV of subgroup II (HTLV-II) was first isolated from a patient with a T-cell variant of hairy cell leukemia. To date, this is the only reported isolate of HTLV-II from a patient with a neoplastic disease. Virus isolation and seroepidemiological data show that HTLV of both subgroups can sometimes be found in patients with AIDS.
Evidence suggests that the retrovirus(es) of the HTLV family is an etiological agent of AIDS based on the following: (1) there is precedence for an animal retrovirus cause of immune deficiency (feline leukemia virus in cats); (2) retroviruses of the HTLV family are T-cell tropic; (3) they preferentially infect "helper" T-cells (OKT4.sup.+); (4) they have cytopathic effects on various human and mammalian cells as demonstrated by their induction of cell syncytia formation; (5) they can alter some T-cell functions; (6) in some cases infection may result in selective T-cell killing; and (7) they are transmitted by intimate contact or through blood products. The presence of antibodies directed to cell membrane antigens of HTLV infected cells has been shown in sera of more than 40% of patients with AIDS [Essex et al., Science, 220:859 (1983)]. This antigen has since been defined as part of the envelope of HTLV [Schupbach, et al., Science, in press; and Lee, et al., Proc. Nat. Acad. Sci. U.S.A., in press].
The original detection and isolation of the various HTLV isolates were made possible by two earlier developments: the discovery of T-cell growth factor (TCGF), also called Interleukin 2 (Il-2), which enabled the routine selective growth of different subsets of normal and neoplastic mature T-cells [Ruscetti, et al., J. Immunol., 119:131 (1977); and Poiesz, et al., Proc. Nat. Acad. Sci. U.S.A, 77:6134 (1980)] and the development of sensitive assays for detection of retroviruses based on reverse transcriptase assays. The methods of HTLV isolation and transmission involved a cocultivation procedure using permissive T-cells for the virus. The use of normal human T-cells in cocultivation experiments preferentially yielded HTLV of both subgroups with immortalizing (transforming) capability for some of the target T-cells.
However, HTLV variants (now termed HTLV-III), lack immortalizing properties for normal T-cells and mainly exhibit cytopathic effects on the T-cells and is now believed to be the cause of AIDS. In fact, such variants were frequently but only transiently detected using these normal T-cells as targets in cocultivation or cell-free transmission experiments. The cytopathic effect was overcome by finding a highly susceptible, permissive cell for cytopathic variants of HTLV, thus preserving the capacity for permanent growth after infection with the virus. The present invention discloses the identification and characterization of this new immortalized T-cell population and its use in the isolation and continuous high- level production of such viruses from patients with AIDS and pre-AIDS.
Early experiments identified one neoplastic aneuploid T-cell line, termed HT, derived from an adult with lymphoid leukemia, that was susceptible to infection with the new cytopathic virus isolates.
This cell line is a sensitive target for transmission of these virus isolates (HTLV-III) and it allows continuous large-scale virus production and development of specific immunologic reagents and nucleic acid probes useful for comparison of these new isolates among themselves and with HTLV-I and HTLV-II. In addition to their differences in biological effects that distinguish them from HTLV-I and HTLV-II, HTLV-III also differs from these known HTLV subgroups in several immunological assays and in morphology. However, these new retroviruses are T4 lymphotropic and exhibit many properties similar to HTLV-I and II, including similar properties of the reverse transcriptase, cross reactivity of structural proteins as determined by heterologous competition radioimmune assays with patients' sera and with animal hyperimmune sera, and induction of syncytia. These new retrovirus isolates are collectively designated HTLV-III, together with detectable differences in some of their proteins and genetic information, HTLV-III's ability to kill T-cells clearly separates these variants from other members of the HTLV family.
STATEMENT OF DEPOSIT
A cell line corresponding to the present invention, and denoted H9/HTLV-III.sub.B, has been deposited in the ATCC (under ATCC No. CRL 8543) on April 19, 1984, prior to the filing of this patent application. This deposit assures permanence of the deposit and ready accessibility thereto by the public. H9 is a representative and preferred cell line in accordance with the invention.
UTILITY STATEMENT
The cell line which is a product of the present invention (H9/HTLV-III.sub.B) is presently useful for the production of vaccines for the relief of AIDS and for the detection of antibodies to the virus in blood samples.
GENERAL DESCRIPTION
A susceptible cell line HT was tested for HTLV before in vitro infection and it was negative by all criteria, including lack of proviral sequences. Continuous production of HTLV-III is obtained after repeated exposure of parental HT cells (3.times.10.sup.6 cells pretreated with polybrene) to concentrated culture fluids containing HTLV-III harvested from short term cultured T-cells (grown with TCGF) which originated from patients with pre-AIDS or AIDS. The concentrated fluids were first shown to contain particle associated reverse transcriptase (RT). When cell proliferation declined, usually 10 to 20 days after exposure to the culture fluids, the fresh (uninfected) HT parental cells are added to cultures. Culture fluids from the infected parental cell line was positive for particulate RT activity and about 20% of the infected cell population was positive in an indirect immune fluorescent assay (IFA) using serum from a hemophilia patient with pre-AIDS (patient E.T.). Serum from E.T. also contained antibodies to proteins of disrupted HTLV-III but did not react with proteins of HTLV-I or HTLV-II infected cells.
SPECIFIC DISCLOSURE
As has been mentioned above, an aneuploid HT-cell line exhibited the desired prerequisites for the continuous propagation of HTLV-III. This cell line is a neoplastic aneuploid T-cell line derived from an adult patient with lymphoid leukemia, selected for its mature T-cell phenotype [OKT3.sup.+ (62%), OKT.sup.4 + (39%) and OKT8.sup.- ], as determined by cytofluorometry using a fluorescence-activated cell sorter. Cultures of these cells are routinely maintained in RPMI/1640 with 20% fetal calf serum and antibiotics. These cultures are shown in Example 1, Table 1. Clone H9 is preferred, with Clone H4 being secondarily preferred.
HTLV-III culture fluids are isolated from cultured cells of patients with acquired immune deficiency syndrome (AIDS). Peripheral blood leukocytes from these patients are banded in Ficoll-Hypaque, incubated in growth media (RPMI 1640, 20% fetal bovine serum 0.29 mg/ml glutamine) containing 5 .mu.g/ml phytohemagglutinin (PHA-P) for 48 hours, at 37.degree. C. in a 5% CO.sub.2 atmosphere. The leukocytes are then refed with growth medium containing 10% purified T cell growth factor (TCGF); optionally, some of the cells also received rabbit antibody to alpha interferon. Cells and growth media from these lymphocytes are then assayed for the presence of HTLV subgroups I-III. Samples exhibiting more than one of the following were considered positive: repeated detection of a Mg.sup.++ dependent reverse transcriptase activity in supernatant fluids; virus observed by electron microscopy; intracellular expression of virus-related antigens detected with antibodies from sero-positive donors or with hyperimmune serum; or transmission of particles, detected by reverse transcriptase assays or by electron microscopic observation, to fresh human cord blood, bone marrow, or peripheral blood T-lymphocytes. All isolates not classified as either HTLV-I or HTLV-II by immunological or nucleic acid analysis were classified as HTLV-III. The cells in the HTLV-III producing cell cultures, characterized using established immunological procedures, are predominantly T-lymphocytes (E rosette receptor, OKT/3 and Leu/1 positive, with a T4 phenotype (OKT4, leu 3a positive). This process is also described by Gallo, et al., in Science, 220:865-867 (1983).
The infection of parental HT cells as well as other cloned cell populations occurs by exposure of these cells to concentrated or nonconcentrated culture fluids (cell-free infection) from T-cell cultures from AIDS or pre-AIDS patients, or by cocultivation; that is, HT cells are infected by exposure to HTLV-III containing cultures. The usual cell-free infection procedure is as follows: 2 to 5.times.10.sup.6 cells are treated with polybrene (2 .mu.g/ml) or DEAE dextran for 30 minutes in CO.sub.2 incubator at 37.degree. C., and then exposed to the virus inoculum (0.1 to 1 ml) for one hour in the incubator (CO.sub.2 /37.degree. C.). The cells are kept in suspension by shaking at regular intervals. After one hour of incubation a regular growth medium is added. The positivity of infected cultures for HTLV-III is assessed after one, two, and three weeks of cultivation.
The infection of HT cells (clones) is also obtained by cocultivation procedure--HT cells are mixed in various proportions (usually 1:5) with short- term cultured T-cells (about 5 to 20 days) from AIDS or pre-AIDS patients. The positivity for HTLV-III was scored by the detection of viral antigens or viral nucleic acid sequences in the infected recipient cells at various intervals (7, 14, 21 days, etc.) after cocultivation. The mixed cultures are maintained in growth medium for several months.
EXAMPLE 1
As shown in Table 1 below, single cell HT clones were isolated as described by Popovic, et al., in Neoplasma, 18:257 (1971), and Bach, et al., Immunol. Rev., 54:5 (1981) from a long-term cultured aneuploid HT-cell line exhibiting mature T-cell phenotype (OKT3.sup.+ [62%], OKT4.sup.+ [39%] and OKT8.sup.-) as determined by cytofluorometry using a fluorescence-activated cell sorter. The cultures were routinely maintained in RPMI/1640 with 20% fetal calf serum and antibiotics. The terminal cell density of the parental cell culture, seeded at a concentration of 2.times.10.sup.5 cells/milliliter of culture media, was in the range 10.sup.6 -1.5.times.10.sup.6 cells/ml after 5 days of culture.
For detection of multinucleated cells, cell smears were prepared from cultures 6 and 14 days after infection and stained with Wright-Giemsa. Cells having more than 5 nuclei were considered as multi-nucleated cells. Cloned cells from uninfected cultures also contained some multi-nucleated giant cells as well; however, the arrangement of multiple nuclei in a characteristic ring formation was lacking and the number of these cells was much less (0.7% to 10%).
Immunofluorescence positive cells were washed with phosphate-buffered saline (PBS) and resuspended in the same buffer at concentration 10.sup.6 cells per milliliter. Approximately 50 .lambda. of cell suspension were spotted on slides, air dried, and fixed in acetone for 10 min. at room temperature. Slides were stored at -20.degree. C. until use. Twenty microliters of either hyperimmune rabbit antiserum to HTLV-III (diluted 1/2000 in PBS) or serum from the patient (E.T.) diluted 1/8 in PBS was applied to cells and incubated for 50 min. at 37.degree. C. The fluorescein-conjugated antiserum to rabbit or human immunogloblin G was diluted and applied to the fixed cells for 30 min. at room temperature. Slides then were washed extensively before microscopic examinations. The uninfected parental cell line as well as the clones were consistently negative in these assays.
To determine reverse transcriptase activity, virus particles were precipitated from cell-free supernatant as follows: 0.4 ml of 4M NaCl and 3.6 ml of 30% (wt/vol.) polyethylene glycol (Carbowax 6000) were added to 8 ml of harvested culture fluids and the suspension was placed on ice overnight. The suspension was centrifuged in a Sorvall RC-3 centrifuge at 2000 rpm at 4.degree. C. for 30 min. The precipitate was resuspended in 300 .mu.l at 50% (vol/vol) glycerol (25 mM Tris-HCl, pH 7.5/5 mM dithiothreitol/150 mM KCl/0.025% Triton X-100. Particles were disrupted by addition of 100 .mu.l of 0.9% Triton X-100/1.5M KCl. Reverse transcriptase (RT) assays were performed as described by Poiesz, et al., Proc Nat. Acad. Sci. U.S.A., 77:7415 (1980) and expressed in cpm per milliliter culture medium.
TABLE 1 __________________________________________________________________________ Response of Cloned T-Cell Populations to HTLV-III Infection Clones Characteristics H3 H4 H6 H9 H17 H31 H35 H38 __________________________________________________________________________ Total cell number (.times. 10.sup.6) 6 days after infection 1 1.5 1.5 0.3 0.4 0.3 0.5 1.8 14 days after infection 2.2 7.3 7.5 10.0 4.7 5.0 4.5 3.2 Multinucleated cells (%) 6 days after infection 24 42 32 7 13 14 30 45 14 days after infection 45 48 45 30 22 45 60 60 Immunofluorescence positive cells (%) 6 days after infection Rabbit antiserum to HTLV-III 55 56 32 32 39 21 10 87 Patient serum (E.T.) 56 29 21 ND ND ND ND 73 14 days after infection Rabbit antiserum to HTLV-III 50 74 60 97 71 40 20 80 Patient serum 45 47 56 78 61 43 22 89 Reverse transcriptase activity (.times. 10.sup.4 cpm/ml) 6 days after infection 2.4 1.8 2.1 4.1 2.6 1.4 1.7 2.5 14 days after infection 16.2 18.1 16.1 20.2 17.1 13.4 15.1 18.2 __________________________________________________________________________ ND = not done
EXAMPLE 2
As shown in Table 2 below, cocultivation with H4 recipient T-cell clone was performed with fresh mononuclear cells from peripheral blood of patients RF and SN, respectively. In the case of patients BK and LS cocultivation was performed with T-cells grown in the presence of exogenous TCGF (10% V/V) for 10 days. The ratio recipient/donor (patients') cells was 1:5. The mixed cultures were maintained in RPMI/1640 (20% FCS and antibiotics) in the absence of exogenous TCGF. Cell-free infection of H9 T-cell clone was performed using concentrated culture fluids harvested from T-cell cultures of the patient WT. The T-cell cultures were grown in the presence of exogenous TCGF for two weeks before the culture fluids were harvested and concentrated. Cells of H9 clones were pretreated with polybrene (2 .mu.g/ml) for 20 min. and 2.times.10.sup.6 cells were exposed for one hr. to 0.5 ml of 100-fold concentrated culture fluids positive for particulate RT activity.
HTLV-III virus expression in both cocultured and cell-free infected cell cultures were assayed approximately one month after in vitro cultivation. There was considerable fluctuation in HTLV-III expression (see Table 2). For details of both reverse transcriptase (RT) assays and indirect immunofluorescence assays (IFA) see Example 1.
TABLE 2 __________________________________________________________________________ Isolation of HTLV-III from Patients with Pre-AIDS and AIDS Virus Expression IFA with RT Activity Rabbit Serum Human Serum (ET) Patient Diagnosis Origin (.times. 10 cpm) (% Positive) (% Positive) EM __________________________________________________________________________ RF AIDS Haiti 0.25 80 33 ND (heterosexual) SN Hemophiliac U.S. 6.3 10 ND + (lymphadenopathy) BK AIDS U.S. 0.24 44 5 + (homosexual) LS AIDS U.S. 0.13 64 19 + (homosexual) WT Hemophiliac U.S. 3.2 69 ND ND (lymphadenopathy) __________________________________________________________________________ RT = reverse transcriptase IFA = immunofluorescence assays EM = electron microscopy ND = not done
EXAMPLE 3
To select for high permissiveness for HTLV-III and to preserve permanent growth and continuous production of virus, extensive cloning of the HT parental T-cell population was performed. A total of 51 single-cell clones was obtained by both capillary and limited dilution techniques using irradiated mononuclear cells from peripheral blood of a healthy donor as a feeder. The growth of these cell clones was compared after HTLV-III infection. A representative example of response to virus infection of 8 T-cell clones which are susceptible to and permissive for HTLV-III is shown in Table 1. In parallel experiments, 2.times.10.sup.6 cells of each T-cell clone were exposed to 0.1 ml of concentrated virus. Then cell growth and morphology, expression of cellular viral antigen(s), and RT activity in culture fluids were assessed 6 and 14 days after infection. Although all 8 clones were susceptible to and permissive for the virus, there were considerable differences in their ability to proliferate after infection. The cell number decreased by 10% to 90% from the initial cell count within 6 days after infection, and a high proportion of multinucleated (giant) cells were consistently found in all 8 infected clones. The percentage of T-cells positive for viral antigen(s) determined by immunofluorescent assays with serum from AIDS patient (E.T.) and with hyperimmune rabbit serum raised against the whole disrupted HTLV-III ranged from 10% to over 80%. Fourteen days after infection, the total cell number and the proportion of HTLV-III positive cells increased in all 8 clones. The virus positive cultures consistently showed round giant cells which contained numerous nuclei. These multinucleataed giant cells are similar to those induced by HTLV-I and HTLV-II except that the nuclei exhibit a characteristic ring formation. Electron microscopic examinations showed that the cells released considerable amounts of virus.
EXAMPLE 4
To determine whether HTLV-III is continuously produced by the infected T-cells in long-term cultures, both virus production and cell viability of the infected clone, H4, were followed for several months. Although the virus production fluctuated, culture fluids harvested from the H4/HTLV-III cell cultures at approximately 14-day intervals consistently exhibited particulate RT activity which has been followed for over 5 months. The viability of the cells ranged from 65% to 85% and doubling time of the cell population, which is called H4/HTLV-III, was approximately 30-40 hours. Thus, this permanently growing T-cell population can continuously produce HTLV-III.
The yield of virus produced by H4/HTLV-III cells was assessed by purification of concentrated culture fluids through a sucrose density gradient and assays of particulate RT activity in each fraction collected from the gradient. The highest RT activity was found at density 1.16 g/ml, similar to other retroviruses.
DEPOSIT STATEMENT
[0001] The invention provides the isolated human Ebola (hEbola) viruses denoted as Bundibugyo (EboBun) deposited with the Centers for Disease Control and Prevention ("CDC";
Atlanta, Georgia, United States of America) on November 26, 2007 and accorded an accession number 200706291. This deposit was not made to an International Depository Authority (IDA) as established under the Budapest Treaty on the International Recognition of the Deposit of Microorganisms for the Purposes of Patent Procedure, and is a non-Budapest treaty deposit. The deposited organism is not acceptable by American Type Culture Collection (ATCC), Manassas, Virginia, an International Depository Authority (IDA) as established under the Budapest Treaty on the International Recognition of the Deposit of Microorganisms for the Purposes of Patent Procedure. Samples of the stated Deposit Accession No. 200706291 will be made available to approved facilities for thirty years from the date of deposit, and for the lifetime of the patent issuing from, or claiming priority to this application.
RELATED APPLICATIONS
[0002] This application claims priority benefit of U.S. Provisional Application 61/108,175 filed 24 October 2008; the contents of which are hereby incorporated by reference.
FIELD OF THE INVENTION
[0003] The invention is related to compositions and methods directed to a novel species of human Ebola (hEbola) virus.
BACKGROUND OF THE INVENTION
[0004] The family Filoviridae consists of two genera, Marburgvirus and Ebolavirus, which have likely evolved from a common ancestor'. The genus Ebolavirus includes four species: Zaire, Sudan, Reston and Cote d'Ivoire (Ivory Coast) ebolaviruses, which have, with the exception of Reston and Cote d'Ivoire ebolaviruses, been associated with large hemorrhagic fever (HF) outbreaks in Africa with high case fatality (53-90%)2.
[0005] Viruses of each species have genomes that are at least 30-40% divergent from one another, a level of diversity that presumably reflects differences in the ecologic niche they occupy and in their evolutionary history. Identification of the natural reservoir of ebolaviruses remains somewhat elusive, although recent PCR and antibody data suggest that three species of arboreal fruit bats may be carriers of Zaire ebolavirus3. No data has yet been published to suggest reservoirs for the Sudan, Reston and Cote d'Ivoire ebolavirus species. However, a cave-dwelling fruit bat has been recently implicated as a natural host for marburgvirus4' s, supporting the hypothesis that different bat species may be the reservoir hosts for the various filoviruses.
[0006] Filovirus outbreaks are sporadic, sometimes interspersed by years or even decades of no apparent disease activity. The last new species of ebolavirus was discovered 14 years ago (1994), in Cote d'Ivoire (Ivory Coast), and involved a single non-fatal case, a veterinarian who performed an autopsy on an infected chimpanzee found in the Tai Forest6. No further disease reports have been associated with Cote d'Ivoire ebolavirus, in contrast to Zaire and Sudan ebolaviruses which have each caused multiple large outbreaks over the same time period.
[0007] In late November 2007, HF cases were reported in the townships of Bundibugyo and Kikyo in Bundibugyo District, Western Uganda. The outbreak continued through January 2008, and resulted in approximately 149 cases and 37 deaths. Laboratory investigation of the initial 29 suspect-case blood specimens by classic methods (antigen capture, IgM and IgG
ELISA) and a recently developed random-primed pyrosequencing approach identified this to be an Ebola HF
outbreak associated with a new discovered ebolavirus species. These specimens were negative when initially tested with highly sensitive real-time RT-PCR assays specific for all known Zaire and Sudan ebolaviruses and Marburg viruses. This new species is referred to herein as "the Bundibugyo species", abbreviated "EboBun".
[0008] Accordingly, compositions and methods directed to the new Ebola virus species are described herein and the most closely related Ebola Ivory Coast species, which compositions and methods are useful for diagnosis and prevention of human Ebola virus infection; including related vaccine development, and prevention of hemorrhagic fever in a human population.
SUMMARY OF THE INVENTION
[0009] The present invention is based upon the isolation and identification of a new human Ebola virus species, EboBun. EboBun was isolated from the patients suffering from hemorrhagic fever in a recent outbreak in Uganda. The isolated virus is a member of the Filoviridae family, a family of negative sense RNA viruses. Accordingly, the invention relates to the isolated EboBun virus that morphologically and phylogenetically relates to known members filoviridae.
[0010] In one aspect, the invention provides the isolated EboBun virus deposited with the Centers for Disease Control and Prevention ("CDC"; Atlanta, Georgia, United States of America) on November 26, 2007 and accorded an accession number 200706291, as stated in the paragraph entitled "DEPOSIT STATEMENT" supra.
[0011] In another aspect, the invention provides an isolated hEbola EboBun virus comprising a nucleic acid molecule comprising a nucleotide sequence selected from the group consisting of: a) a nucleotide sequence set forth in SEQ ID NO: 1; b) a nucleotide sequence that hybridizes to the sequence set forth in SEQ ID NO: 1 under stringent conditions; and c) a nucleotide sequence that has at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to the SEQ ID NO:
1. In another aspect, the invention provides the complete genomic sequence of the hEbola virus EboBun.
[0012] In a related aspect, the invention provides nucleic acid molecules isolated from EboBun, or fragments thereof.
[0013] In another aspect, the invention provides proteins or polypeptides that are isolated from the EboBun, including viral proteins isolated from cells infected with the virus but not present in comparable uninfected cells; or fragments thereof. In one embodiment of the present invention, the amino acid sequences of the proteins or polypeptides are set forth in SEQ ID
NOS: 2-9 and 59, or fragments thereof.
[0014] In a related aspect, the invention provides an isolated polypeptide encoded by the nucleic acid molecule of the inventive hEbola EboIC (Sequence ID No. 10) virus described above.
[0015] In another aspect, the invention provides an isolated hEbola EboIC
virus comprising a nucleic acid molecule comprising a nucleotide sequence selected from the group consisting of: a) a nucleotide sequence set forth in SEQ ID NO: 10; b) a nucleotide sequence that hybridizes to the sequence set forth in SEQ ID NO: 10 under stringent conditions; and c) a nucleotide sequence that has at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to the SEQ ID NO:
10. In another aspect, the invention provides the complete genomic sequence of the hEbola virus EboIC.
[0016] In a related aspect, the invention provides nucleic acid molecules isolated from EboIC, or fragments thereof.
[0017] In another aspect, the invention provides proteins or polypeptides that are isolated from the EboIC, including viral proteins isolated from cells infected with the virus but not present in comparable uninfected cells; or fragments thereof. In one embodiment of the present invention, the amino acid sequences of the proteins or polypeptides are set forth in SEQ ID
NOs: 11-19, or fragments thereof.
[0018] In a related aspect, the invention provides an isolated polypeptide encoded by the nucleic acid molecule of the inventive hEbola EboIC virus described above.
[0019] In other aspects, the invention relates to the use of the isolated hEbola virus for diagnostic and therapeutic methods based on EbBun, EboIC, or a combination thereof. In one embodiment, the invention provides a method of detecting in a biological sample an antibody immunospecific for the genus of West Afrin Ebola Species constituting hEbola EbBun and EboIC
virus using at least one the inventive isolated hEbola virus described herein, or any of the inventive proteins or polypeptides as described herein. In another specific embodiment, the invention provides a method of screening for an antibody which immunospecifically binds and neutralizes hEbola EboBun. Such an antibody is useful for a passive immunization or immunotherapy of a subject infected with hEbola.
[0020] In another aspect, the invention provides an isolated antibody or an antigen-binding fragment thereof which immunospecifically binds to the hEbola virus of the invention described above.
[0021] In other aspects, the invention provides methods for detecting the presence, activity or expression of the Glade of Bundibungyo-Ivory Coast hEbola virus in a biological material, such as cells, blood, saliva, urine, feces and so forth; and specifically at least one of EbBun or EboIC.
[0022] In a related aspect, the invention provides a method for detecting the presence of the inventive hEbola virus described above in a biological sample, the method includes (a) contacting the sample with an agent that selectively binds to a West African hEbola virus; and (b) detecting whether the compound binds to the West African hEbola virus in the sample.
[0023] In another aspect, the invention provides a method for detecting the presence of the inventive polypeptide described above, in a biological sample, said method includes (a) contacting the biological sample with an agent that selectively binds to the polypeptide;
and (b) detecting whether the agent binds to the polypeptide in the sample. In another aspect, the invention provides a method for detecting the presence of a first nucleic acid molecule derived from the inventive hEbola virus described above in a biological sample, the method comprising: (a) contacting the biological sample with an agent that selectively binds to the polypeptide; and (b) detecting whether the agent binds to the polypeptide in the sample.
[0024] In another aspect, the invention provides a method for propagating the hEbola virus in host cells comprising infecting the host cells with the inventive isolated hEbola virus described above, culturing the host cells to allow the virus to multiply, and harvesting the resulting virions.
Also provided by the present invention are host cells infected with the inventive hEbola virus 5 described above.
[0025] In another aspect, the invention provides a method of detecting in a biological sample the presence of an antibody that immunospecifically binds hEbola virus, the method comprising: (a) contacting the biological sample with the inventive host cell host described above; and (b) detecting the antibody bound to the cell.
[0026] In another aspect, the invention provides vaccine preparations, comprising the inventive hEbola virus, including recombinant and chimeric forms of the virus, nucleic acid molecules comprised by the virus, or protein subunits of the virus. The invention also provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of the inventive hEbola virus described above, and a pharmaceutically acceptable carrier. In one embodiment, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a protein extract of the inventive hEbola virus described above, or a subunit thereof; and a pharmaceutically acceptable carrier. In another, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NO: 1 or a complement thereof, and a pharmaceutically acceptable carrier. In another, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule comprising any of inventive the nucleotide sequences as described above, or a complement thereof, and a pharmaceutically acceptable carrier.
[0027] In a related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of the inventive hEbola virus described above, and a pharmaceutically acceptable carrier. In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of a protein extract of the inventive hEbola virus described above or a subunit thereof, and a pharmaceutically acceptable carrier. In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NO: 1 or a complement thereof, and a pharmaceutically acceptable carrier. In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of a nucleic acid molecule comprising the inventive nucleotide sequence as described above or a complement thereof, and a pharmaceutically acceptable carrier. In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of any of the inventive polypeptides described above.
[0028] In another aspect, the present invention provides pharmaceutical compositions comprising antiviral agents of the present invention and a pharmaceutically acceptable carrier. In a specific embodiment, the antiviral agent of the invention is an antibody that immunospecifically binds hEbola virus or any hEbola epitope. In another specific embodiment, the antiviral agent is a polypeptide or protein of the present invention or nucleic acid molecule of the invention.
[0029] In a related aspect, the invention provides a pharmaceutical composition comprising a prophylactically or therapeutically effective amount of an anti-hEbola EboBun agent and a pharmaceutically acceptable carrier.
[0030] The invention also provides kits containing compositions and formulations of the present invention. Thus, in another aspect, the invention provides a kit comprising a container containing the inventive immunogenic formulation described above. In another aspect, the invention provides a kit comprising a container containing the inventive vaccine formulation described above. In another, the invention provides a kit comprising a container containing the inventive pharmaceutical composition described above. In another, the invention provides a kit comprising a container containing the inventive vaccine formulation described above. In another, the invention provides a method for identifying a subject infected with the inventive hEbola virus described above, comprising: (a) obtaining total RNA from a biological sample obtained from the subject; (b) reverse transcribing the total RNA to obtain cDNA; and (c) amplifying the cDNA using a set of primers derived from a nucleotide sequence of the inventive hEbola virus described above.
[0031] The invention further relates to the use of the sequence information of the isolated virus for diagnostic and therapeutic methods.
[0032] In another aspect, the present invention provides methods for screening antiviral agents that inhibit the infectivity or replication of hEbola virus or variants thereof.
[0033] The invention further provides methods of preparing recombinant or chimeric forms of hEbola.
BRIEF DESCRIPTION OF THE DRAWINGS
[0034] FIG. 1 represents a Phylogenetic tree comparing full-length genomes of Ebolavirus and Marburg virus by Bayesian analysis;
[0035] FIG. 2 represents an alignment of genomes of novel hEbola EboBun (SEQ
ID NO: 1) referred to below as "Ebola Bundibugyo" or "EboBun", and hEbola Zaire (SEQ ID
NO: 20);
referred to below as "Ebola Zaire `76" or "EboZ" and hEbola Ivory Coast (SEQ
ID NO: 10) also referred to below as "EboIC".
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
[0036] It is to be understood that the present invention is not limited to particular embodiments described, as such may, of course, vary. It is also to be understood that the terminology used herein is for the purpose of describing particular embodiments only, and is not intended to be limiting.
[0037] Due to the sequence divergence of EboBun relative to all previously recognized ebolaviruses, the present invention has utility in design of diagnostic assays to monitor Ebola HF
disease in humans and animals, and develop effective antivirals and vaccines.
[0038] The EboBun virus of the present invention is genetically distinct, differing by more than 30% at the genome level from all other known ebolavirus species. The unique nature of this virus created challenges for traditional filovirus molecular based diagnostic assays and genome sequencing approaches. Instead, over 70% of the virus genome was sequenced using a recently developed random-primed pyrosequencing approach which allowed the rapid development of molecular detection assay which were deployed in the disease outbreak response. This random-primed pyrosequencing draft sequence allowed faster completion of the whole genome sequence using traditional primer walking approach and confirmation that the EboBun virus represented a new ebolavirus species.
Definitions [0039] The definitions herein provided are operative throughout the entire description of the invention set forth herein, including the Summary of the Invention.
[0040] The term "an antibody or an antibody fragment that immunospecifically binds a polypeptide of the invention" as used herein refers to an antibody or a fragment thereof that immunospecifically binds to the polypeptide encoded by the nucleotide sequence of SEQ ID NO: 1 (EboBun), or a fragment thereof, and does not non-specifically bind to other polypeptides. An antibody or a fragment thereof that immunospecifically binds to the polypeptide of the invention may cross-react with other antigens. Preferably, an antibody or a fragment thereof that immunospecifically binds to a polypeptide of the invention does not cross-react with other antigens.
An antibody or a fragment thereof that immunospecifically binds to the polypeptide of the invention can be identified by, for example, immunoassays or other techniques known to those skilled in the art, or otherwise as described herein.
[0041] An "isolated" or "purified" peptide or protein is substantially free of cellular material or other contaminating proteins from the cell or tissue source from which the protein is derived, or substantially free of chemical precursors or other chemicals when chemically synthesized. The language "substantially free of cellular material" includes preparations of a polypeptide/protein in which the polypeptide/protein is separated from cellular components of the cells from which it is isolated or recombinantly produced. Thus, a polypeptide/protein that is substantially free of cellular material includes preparations of the polypeptide/protein having less than about 30%, 20%, 10%, 5%, 2.5%, or 1% (by dry weight) of contaminating protein. When the polypeptide/protein is recombinantly produced, it is also preferably substantially free of culture medium, i.e., culture medium represents less than about 20%, 10%, or 5% of the volume of the protein preparation.
When polypeptide/protein is produced by chemical synthesis, it is preferably substantially free of chemical precursors or other chemicals, i.e., it is separated from chemical precursors or other chemicals which are involved in the synthesis of the protein. Accordingly, such preparations of the polypeptide/protein have less than about 30%, 20%, 10%, 5% (by dry weight) of chemical precursors or compounds other than polypeptide/protein fragment of interest.
In a preferred embodiment of the present invention, polypeptides/proteins are isolated or purified.
[0042] An "isolated" nucleic acid molecule is one which is separated from other nucleic acid molecules which are present in the natural source of the nucleic acid molecule. Moreover, an "isolated" nucleic acid molecule, such as a cDNA molecule, can be substantially free of other cellular material, or culture medium when produced by recombinant techniques, or substantially free of chemical precursors or other chemicals when chemically synthesized. In a preferred embodiment of the invention, nucleic acid molecules encoding polypeptides/proteins of the invention are isolated or purified. The term "isolated" nucleic acid molecule does not include a nucleic acid that is a member of a library that has not been purified away from other library clones containing other nucleic acid molecules.
[0043] The term "portion" or "fragment" as used herein includes the specified fragment lengths, and all integers in between, inclusive of the specified end points in a specified range, and inclusive of any length up to the full length of a protein, polypeptide, or nucleic acid.
[0044] The term "having a biological activity of the protein" or "having biological activities of the polypeptides of the invention" refers to the characteristics of the polypeptides or proteins having a common biological activity, similar or identical structural domain, and/or having sufficient amino acid identity to the polypeptide encoded by the nucleotide sequence of SEQ ID
NO: 1 (EboBun).
Such common biological activities of the polypeptides of the invention include antigenicity and immunogenicity.
[0045] The term "under stringent condition" refers to hybridization and washing conditions under which nucleotide sequences having at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, or at least 95% identity to each other remain hybridized to each other. Such hybridization conditions are described in, for example but not limited to, Current Protocols in Molecular Biology, John Wiley & Sons, NY (1989), 6.3.1-6.3.6.; Basic Methods in Molecular Biology, Elsevier Science Publishing Co., Inc., NY (1986), pp. 75-78, and 84-87; and Molecular Cloning, Cold Spring Harbor Laboratory, NY (1982), pp. 387-389, and are well known to those skilled in the art. A preferred, non-limiting example of stringent hybridization conditions is hybridization in 6 x sodium chloride/sodium citrate (SSC), 0.5% SDS at about 68 C followed by one or more washes in 2 x SSC, 0.5% SDS at room temperature. Another preferred, non-limiting example of stringent hybridization conditions is hybridization in 6 x SSC at about 45 C, followed by one or more washes in 0.2 x SSC, 0.1% SDS at about 50-65 C.
[0046] The term "variant" as used herein refers either to a naturally occurring genetic mutant of hEbola EboBun, or hEbola EboIC, or a recombinantly prepared variation of these hEbola species, each of which contain one or more mutations in its genome compared to the hEbola of SEQ ID NO:
1 or 10. The term "variant" may also refer either to a naturally occurring variation of a given peptide or a recombinantly prepared variation of a given peptide or protein in which one or more amino acid residues have been modified by amino acid substitution, addition, or deletion.
[0047] "Homology" refers to sequence similarity or, alternatively, sequence identity, between two or more polynucleotide sequences or two or more polypeptide sequences.
[0048] The terms "percent identity" and "% identity," as applied to polynucleotide sequences, refer to the percentage of identical nucleotide matches between at least two polynucleotide sequences aligned using a standardized algorithm. Such an algorithm may insert, in a standardized and reproducible way, gaps in the sequences being compared in order to optimize alignment between two sequences, and therefore achieve a more meaningful comparison of the two sequences.
[0049] Percent identity between polynucleotide sequences may be determined using one or more computer algorithms or programs known in the art or described herein. For example, percent 5 identity can be determined using the default parameters of the CLUSTAL V
algorithm as incorporated into the MEGALIGN version 3.12e sequence alignment program. This program is part of the LASERGENE software package, a suite of molecular biological analysis programs (DNASTAR, Madison, Wis.). CLUSTAL V is described in Higgins, D. G. and P. M.
Sharp (1989;
CABIOS 5:151-153) and in Higgins, D. G. et al. (1992; CABIOS 8:189-191). For pairwise 10 alignments of polynucleotide sequences, the default parameters are set as follows: Ktuple=2, gap penalty=5, window=4, and "diagonals saved"=4. The "weighted" residue weight table is selected as the default.
[0050] Alternatively, a suite of commonly used and freely available sequence comparison algorithms which can be used is provided by the National Center for Biotechnology Information (NCBI) Basic Local Alignment Search Tool (BLAST) (Altschul, S. F. et al.
(1990) J. Mol. Biol.
215:403-410), which is available from several sources, including the NCBI, Bethesda, Md., and on the NCBI World Wide Web site available on the Internet. The BLAST software suite includes various sequence analysis programs including "blastn," that is used to align a known polynucleotide sequence with other polynucleotide sequences from a variety of databases. Also available is a tool called "BLAST 2 Sequences" that is used for direct pairwise comparison of two nucleotide sequences. "BLAST 2 Sequences" can be accessed and used interactively on the Internet via the NCBI World Wide Web site as well. The "BLAST 2 Sequences" tool can be used for both blastn and blastp (discussed below). BLAST programs are commonly used with gap and other parameters set to default settings. For example, to compare two nucleotide sequences, one may use blastn with the "BLAST 2 Sequences" tool Version 2Ø12 (Apr. 21, 2000) set at default parameters. Such default parameters may be, for example: Matrix:BLOSUM62; Reward for match: 1;
Penalty for mismatch: -2; Open Gap: 5 and Extension Gap: 2 penalties; Gap x drop-off: 50;
Expect: 10; Word Size: 11; Filter: on.
[0051] Percent identity may be measured over the length of an entire defined sequence, for example, as defined by a particular SEQ ID number, or may be measured over a shorter length, for example, over the length of a fragment taken from a larger, defined sequence, for instance, a fragment of at least 20, at least 30, at least 40, at least 50, at least 70, at least 100, or at least 200 contiguous nucleotides. Such lengths are exemplary only, and it is understood that any fragment length supported by the sequences shown herein, in the tables, figures, or sequence listing, may be used to describe a length over which percentage identity may be measured.
[0052] The phrases "percent identity" and "% identity", as applied to polypeptide sequences, refer to the percentage of identical residue matches between at least two polypeptide sequences aligned using a standardized algorithm. Methods of polypeptide sequence alignment are well known. Some alignment methods take into account conservative amino acid substitutions. Such conservative substitutions, explained in more detail above, generally preserve the charge and hydrophobicity at the site of substitution, thus preserving the structure (and therefore function) of the polypeptide. The phrases "percent similarity" and "% similarity", as applied to polypeptide sequences, refer to the percentage of residue matches, including identical residue matches and conservative substitutions, between at least two polypeptide sequences aligned using a standardized algorithm. In contrast, conservative substitutions are not included in the calculation of percent identity between polypeptide sequences.
[0053] Percent identity between polypeptide sequences may be determined using the default parameters of the CLUSTAL V algorithm as incorporated into the MEGALIGN
version 3.12e sequence alignment program (described and referenced above). For pairwise alignments of polypeptide sequences using CLUSTAL V, the default parameters are set as follows: Ktuple=l, gap penalty=3, window=5, and "diagonals saved"=5. The PAM250 matrix is selected as the default residue weight table.
[0054] Alternatively the NCBI BLAST software suite may be used. For example, for a pairwise comparison of two polypeptide sequences, one may use the "BLAST 2 Sequences"
tool Version 2Ø12 (Apr. 21, 2000) with blastp set at default parameters. Such default parameters may be, for example: Matrix: BLOSUM62; Open Gap: 11 and Extension Gap: 1 penalties; Gap x drop-off: 50;
Expect: 10; Word Size: 3; Filter: on.
[0055] Percent identity may be measured over the length of an entire defined polypeptide sequence, for example, as defined by a particular SEQ ID number, or may be measured over a shorter length, for example, over the length of a fragment taken from a larger, defined polypeptide sequence, for instance, a fragment of at least 15, at least 20, at least 30, at least 40, at least 50, at least 70 or at least 150 contiguous residues. Such lengths are exemplary only, and it is understood that any fragment length supported by the sequences shown herein, in the tables, figures or sequence listing, may be used to describe a length over which percentage identity may be measured.
[0056] The term "agent" encompasses any chemical, biochemical, or biological molecule; such as small molecules, proteins, polypeptides, antibodies, nucleic acid molecules including DNA or RNA, and the like.
Methods and compositions related to the inventive hEbola [0057] The present invention is based upon the isolation and identification of a new human Ebola virus species, EboBun and the sequencing of the only other known West African Ebola species EboIC. EboBun was isolated from the patients suffering from hemorrhagic fever in a recent outbreak in Uganda. The isolated virus is a member of the Filoviridae family, a family of negative sense RNA viruses. Accordingly, the invention relates to the isolated EboBun or EBOIC virus that morphologically and phylogenetically relates to known members filoviridae.
[0058] In another aspect, the invention provides an isolated hEbola virus including a nucleic acid molecule with a nucleotide sequence that is preferably: a) a nucleotide sequence set forth in SEQ ID NO: 1; b) a nucleotide sequence that hybridizes to the sequence set forth in SEQ ID NO: 1 under stringent conditions; or c) a nucleotide sequence that has at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to the SEQ ID NO: 1. In one embodiment of the present invention, the hEbola virus is killed. In another, the virus is attenuated. In another, the infectivity of the attenuated hEbola virus is reduced. In another, the infectivity is reduced by at least 5-fold, 10-fold, 25-fold, 50-fold, 100-fold, 250-fold, 500-fold, or 10,000-fold. In another, the replication ability of the attenuated hEbola virus is reduced. In another, the replication ability of the attenuated virus is educed by at least 5-fold, 10-fold, 25-fold, 50-fold, 100-fold, 250-fold, 500-fold, 1,000-fold, or 10,000-fold. In another, the protein synthesis ability of the attenuated virus is reduced. In another, the protein synthesis ability is reduced by at least 5-fold, 10-fold, 25-fold, 50-fold, 100-fold, 250-fold, 500-fold, 1,000-fold, or 10,000-fold. In another, the assembling ability of the attenuated hEbola virus is reduced. In another, the assembling ability of the attenuated virus is reduced by at least 5-fold, 10-fold, 25-fold, 50-fold, 100-fold, 250-fold, 500-fold, 1,000-fold, or 10,000-fold. In another, the cytopathic effect of the attenuated hEbola virus is reduced. In another, the cytopathic effect is reduced by at least 5-fold, 10-fold, 25-fold, 50-fold, 100-fold, 250-fold, 500-fold, 1,000-fold, or 10,000-fold.
[0059] In another aspect, the invention provides the complete genomic sequence of the hEbola virus EboBun or EboIC. In a specific embodiment, the virus includes a nucleotide sequence of SEQ
ID NOs: 1 or 10, respectively.
[0060] In a related aspect, the invention provides nucleic acid molecules isolated from EboBun, EboIC, or fragments thereof. In one embodiment of the present invention, the isolated nucleic acid molecule includes the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof. In another, the nucleic acid molecule includes a nucleotide sequence having at least 4, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 4600, 4700, 4800, or 4900 contiguous nucleotides of the nucleotide sequence of SEQ ID NO: 1, or a complement thereof;
with the proviso that the nucleotide sequence is not comprised by the nucleotide sequence set forth in SEQ ID NO: 20 (Ebola Zaire nucleotide sequence); or at least 5000, 5500, 5600, 5700, 5800, 5900, 6000, 6100, 6200, 6300, 6400, 6500, or 6600 contiguous nucleotides of the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof. In another embodiment, the isolated nucleic acid molecule includes a nucleotide sequence that encodes the EboBun amino acid sequence of SEQ ID NOs: 2-9 or 59, the EboIC amino acid sequence of SEQ ID NOs: 11-19, or a complement of the nucleotide sequence that encodes the EboBun amino acid sequences of SEQ
ID NOs: 2-9 or 59 or the EboIC amino acid sequences of SEQ ID NOs: 11-19. In another, the isolated nucleic acid molecule hybridizes under stringent conditions to a nucleic acid molecule having the nucleotide sequence of SEQ ID NOs: 1 or 10 or a complement thereof, wherein the nucleic acid molecule encodes an amino acid sequence which has a biological activity exhibited by a polypeptide encoded by the nucleotide sequence of SEQ ID NOs: 1 or 10. In another, nucleic acid molecule is RNA. In another, nucleic acid molecule is DNA.
[0061] In another aspect, the invention provides proteins or polypeptides that are isolated from the EboBun, including viral proteins isolated from cells infected with the virus but not present in comparable uninfected cells. In one embodiment of the present invention, the amino acid sequences of the proteins or polypeptides are set forth in SEQ ID NOs: 2-9, 59, or 11-19, or fragments thereof.
In one embodiment, polypeptides or proteins of the present invention have a biological activity of the protein (including antigenicity and/or immunogenicity) encoded by the sequence of SEQ ID
NOs: 1 or 10. In another, the polypeptides or the proteins of the present invention have a biological activity of at least one protein having the amino acid sequence (including antigenicity and/or immunogenicity) set forth in SEQ ID NOS: 2-9, 59, or 11-19, or a fragment thereof.
[0062] In a related aspect, the invention provides an isolated polypeptide encoded by the nucleic acid molecule of the invention described above. In one embodiment of the present invention, the isolated polypeptide includes the amino acid sequence selected from the group consisting of: a) an amino acid sequence set forth in SEQ ID NO: 2, 3, 4, 5, 6, 7, 8, or 9; 11, 12, 13, 14, 15, 16, 17, 18 or 19; and b) an amino acid sequence that has 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% homology to the amino acid sequence according to a). In another, the isolated polypeptide comprises the amino acid sequence having at least 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 210, 220, 230, 240 or 250 contiguous amino acid residues of the amino acid sequence of SEQ ID NOs: 5 or 18 (VP24); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 210, 220, 230, 240, 250, 260, 270, 280 contiguous amino acid residues of the amino acid sequence of SEQ ID NOs: 6 or 17 (VP30); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 310, or 320 contiguous amino acid residues of the amino acid sequence of SEQ
ID NOs: 8 or 13 (VP40); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 310, 320, 330, or 340 contiguous amino acid residues of the amino acid sequence of SEQ ID NOs: 7 or 12 (VP35); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 310, 320, 330, 340, 350, 360, or 370 contiguous amino acid residues of the amino acid sequence of SEQ ID
NOs: 4 or 15 (SGP); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 310, 320, 330, 340, 350, 360, or 370 contiguous amino acid residues of the amino acid sequence of SEQ ID NOs: 59 or 16 (SSGP); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, 450, 500, 550, 600, 610, 620, 630, 640, 650, 660, or 670 contiguous amino acid residues of the amino acid sequence of SEQ ID NOs: 9 or 14 (GP); 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, 450, 500, 550, 600, 650, 700, 710, 720, or 730 contiguous amino acid residues of the amino acid sequence of SEQ
ID NOs: 3 or 11 (NP); or 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1050, 1100, 1150, 1200, 1250, 1300, 1350, 1400, 1450, 1500, 1550, 1600, 1650, 1700, 1750, 1800, 1850, 1900, 1950, 2000, 2050, 2100, 2150, 2160, 2170, 2180, 2190, or 2200 contiguous amino acid residues of the amino acid sequence of SEQ ID NOs: 2 or 19 (L).
[0063] In other aspects, the invention relates to the use of an isolated West African hEbola virus for diagnostic and therapeutic methods. In one embodiment, the invention provides a method of detecting in a biological sample an antibody immunospecific for the hEbola virus using the inventive isolated hEbola virus described herein, or any of the inventive proteins or polypeptides as described herein. In another specific embodiment, the invention provides a method of screening for an antibody which immunospecifically binds and neutralizes hEbola EboBun or EboIC
or a combination thereof. Such an antibody is useful for a passive immunization or immunotherapy of a subject infected with hEbola.
[0064] In another aspect, the invention provides an isolated antibody or an antigen-binding fragment thereof which immunospecifically binds to a West African genus hEbola virus of the 5 invention described above, and illustratively including EboBun or EboIC. In one embodiment of the present invention, the isolated antibody or an antigen-binding fragment thereof neutralizes a West African genus hEbola virus. In another, the isolated antibody or an antigen-binding fragment thereof immunospecifically binds to the inventive polypeptide described above. The invention further provides antibodies that specifically bind a polypeptide of the invention encoded by the nucleotide 10 sequence of SEQ ID NOs: 1 (EboBun) or 10 (EboIC), a fragment thereof, or encoded by a nucleic acid comprising a nucleotide sequence that hybridizes under stringent conditions to the nucleotide sequence of SEQ ID NOs: 1 (EboBun) or 10 (EboIC) and/or any hEbola EboBun epitope, having one or more biological activities of a polypeptide of the invention. These polypeptides include those shown in SEQ ID NOs: 2-9, 59, and 11-19. Such antibodies include, but are not limited to, 15 polyclonal, monoclonal, bi-specific, multi-specific, human, humanized, chimeric antibodies, single chain antibodies, Fab fragments, F(ab')2 fragments, disulfide-linked Fvs, intrabodies and fragments containing either a VL or VH domain or even a complementary determining region (CDR) that specifically binds to a polypeptide of the invention.
[0065] In other aspects, the invention provides methods for detecting the presence, activity or expression of the hEbola virus of the invention in a biological material, such as cells, blood, saliva, urine, and so forth. The increased or decreased activity or expression of the hEbola virus in a sample relative to a control sample can be determined by contacting the biological material with an agent which can detect directly or indirectly the presence, activity or expression of the hEbola virus.
In one embodiment of the present invention, the detecting agents are the antibodies or nucleic acid molecules of the present invention. Antibodies of the invention can also be used to treat hemorrhagic fever.
[0066] In a related aspect, the invention provides a method for detecting the presence of the inventive hEbola virus described above in a biological sample, the method comprising:
(a) contacting the sample with an agent that selectively binds to the hEbola virus; and (b) detecting whether the compound binds to the hEbola virus in the sample. In one embodiment of the present invention, the biological sample is selected from the group consisting of cells; blood; serum; plasma;
feces; rectal, vaginal and conjunctival swabs In another, the agent that binds to the virus is an antibody. In another, the agent that binds to the virus is a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NO: 1 or a complement thereof. In another, the agent that binds to the virus is a nucleic acid molecule comprising a nucleotide sequence having at least 4, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 4600, 4700, 4800, 4900, 5000, 5500, 5600, 5700, 5800, 5900, 6000, 6100, 6200, 6300, 6400, 6500, or 6600 contiguous nucleotides of the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof.
[0067] In another aspect, the invention provides a method for detecting the presence of the inventive polypeptide described above, in a biological sample, the method comprising:
(a) contacting the biological sample with an agent that selectively binds to the polypeptide; and (b) detecting whether the agent binds to the polypeptide in the sample. In one embodiment of the present invention, the biological sample is selected from the group consisting of cells; blood; serum;
plasma; feces; rectal, vaginal and conjunctival swabs. In another, the agent that binds to the polypeptide is an antibody or an antigen-binding fragment thereof.
[0068] In another aspect, the invention provides a method for detecting the presence of a first nucleic acid molecule derived from the inventive hEbola virus described above in a biological sample, the method includes (a) contacting the biological sample with an agent that selectively binds to the nucleic acid; and (b) detecting whether the agent binds to the nucleotide in the sample. In one embodiment of the present invention, the agent that binds to the first nucleic acid molecule is a second nucleic acid molecule comprising the nucleotide sequence of SEQ ID NO:
1 or a complement thereof. In another, the second nucleic acid molecule comprises at least 4, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 4600, 4700, 4800, 4900, 5000, 5500, 5600, 5700, 5800, 5900, 6000, 6100, 6200, 6300, 6400, 6500, or 6600 contiguous nucleotides of the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof.
[0069] In another aspect, the invention provides a method for propagating the hEbola virus in host cells comprising infecting the host cells with an inventive isolated West African hEbola virus described above, culturing the host cells to allow the virus to multiply, and harvesting the resulting virions. Also provided by the present invention are host cells infected with the inventive hEbola virus described above. In one embodiment of the present invention, the host cell is a primate cell.
[0070] In another aspect, the invention provides a method of detecting in a biological sample the presence of an antibody that immunospecifically binds hEbola virus, the method includes: (a) contacting the biological sample with the inventive host cell described above;
and (b) detecting the antibody bound to the cell.
[0071] In another aspect, the invention provides vaccine preparations, including the inventive hEbola virus, including recombinant and chimeric forms of the virus, nucleic acid molecules comprised by the virus, or protein subunits of the virus. In one embodiment, the vaccine preparations of the present invention includes live but attenuated hEbola virus with or without pharmaceutically acceptable carriers, including adjuvants. In another, the vaccine preparations of the invention comprise an inactivated or killed hEbola EboBun virus, EboIC
virus, or a combination thereof, with or without pharmaceutically acceptable carriers, including adjuvants. Such attenuated or inactivated viruses may be prepared by a series of passages of the virus through the host cells or by preparing recombinant or chimeric forms of virus. Accordingly, the present invention further provides methods of preparing recombinant or chimeric forms of the inventive hEbola viruses described herein.
[0072] In another specific embodiment, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of the inventive hEbola virus described above, and a pharmaceutically acceptable carrier. In another, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a protein extract of the inventive hEbola virus described above, or a subunit thereof;
and a pharmaceutically acceptable carrier. In another aspect, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof, and a pharmaceutically acceptable carrier. In another, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule comprising any of inventive the nucleotide sequences as described above, or a complement thereof, and a pharmaceutically acceptable carrier. In another aspect, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a protein extract of the inventive hEbola virus described above, or a subunit thereof; and a pharmaceutically acceptable carrier. In another aspect, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof, and a pharmaceutically acceptable carrier. In another, the invention provides a vaccine formulation comprising a therapeutically or prophylactically effective amount of a nucleic acid molecule comprising any of inventive the nucleotide sequences as described above, or a complement thereof, and a pharmaceutically acceptable carrier.
[0073] In yet another specific embodiment, the vaccine preparations of the present invention comprise a nucleic acid or fragment of the hEbola virus, e.g., the virus having Accession No.
200706291, or nucleic acid molecules having the sequence of SEQ ID NOs: 1 or 10, or a fragment thereof. In another, the vaccine preparations comprise a polypeptide of the invention encoded by the nucleotide sequence of SEQ ID NOs: 1 or 10 or a fragment thereof. In a specific embodiment, the vaccine preparations comprise polypeptides of the invention as shown in SEQ ID
NOs: 2-9, 59, or 11-19, or encoded by the nucleotide sequence of SEQ ID NOs: 1 or 10, or a fragment thereof.
[0074] Furthermore, the present invention provides methods for treating, ameliorating, managing or preventing hemorrhagic fever by administering the vaccine preparations or antibodies of the present invention alone or in combination with adjuvants, or other pharmaceutically acceptable excipients. Furthermore, the present invention provides methods for treating, ameliorating, managing, or preventing hemorrhagic fever by administering the inventive compositions and formulations including the vaccine preparations or antibodies of the present invention alone or in combination with antivirals [e.g., amantadine, rimantadine, gancyclovir, acyclovir, ribavirin, penciclovir, oseltamivir, foscamet zidovudine (AZT), didanosine (ddl), lamivudine (3TC), zalcitabine (ddC), stavudine (d4T), nevirapine, delavirdine, indinavir, ritonavir, vidarabine, nelfinavir, saquinavir, relenza, tamiflu, pleconaril, interferons, etc.], steroids and corticosteroids such as prednisone, cortisone, fluticasone and glucocorticoid, antibiotics, analgesics, bronchodilators, or other treatments for respiratory and/or viral infections.
[0075] In a related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of the inventive hEbola virus described above, and a pharmaceutically acceptable carrier.
[0076] In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of a protein extract of the inventive hEbola virus described above or a subunit thereof, and a pharmaceutically acceptable carrier.
[0077] In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NOs: 1, 10, a combination thereof, or a complement thereof, and a pharmaceutically acceptable carrier.
[0078] In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of a nucleic acid molecule comprising the inventive nucleotide sequence as described above or a complement thereof, and a pharmaceutically acceptable carrier.
[0079] In another related aspect, the invention provides an immunogenic formulation comprising an immunogenically effective amount of any of the inventive polypeptides described above.
[0080] In another aspect, the present invention provides pharmaceutical compositions comprising antiviral agents of the present invention and a pharmaceutically acceptable carrier. In a specific embodiment, the antiviral agent of the invention is an antibody that immunospecifically binds hEbola virus or any hEbola epitope. In another specific embodiment, the antiviral agent is a polypeptide or protein of the present invention or nucleic acid molecule of the invention.
[0081] In a related aspect, the invention provides a pharmaceutical composition comprising a prophylactically or therapeutically effective amount of an anti-hEbola EboBun agent and a pharmaceutically acceptable carrier. In one embodiment of the present invention, the anti-hEbola EboBun agent is an antibody or an antigen-binding fragment thereof which immunospecifically binds to the hEbola virus of Deposit Accession No. 200706291, or polypeptides or protein derived therefrom. In another, the anti-hEbola agent is a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NOs: 1, 10, a combination thereof, or a fragment thereof.
In another, the anti-hEbola agent is a polypeptide encoded by a nucleic acid molecule comprising the nucleotide sequence of SEQ ID NOs: 1, 10, a combination thereof, or a fragment thereof having a biological activity of the polypeptide.
[0082] The invention also provides kits containing compositions and formulations of the present invention. Thus, in another aspect, the invention provides a kit comprising a container containing the inventive immunogenic formulation described above.
[0083] In another aspect, the invention provides a kit includes a container containing the inventive vaccine formulation described above.
[0084] In another aspect, the invention provides a kit including a container containing the inventive pharmaceutical composition described above.
[0085] In another aspect, the invention provides a kit including a container containing the inventive vaccine formulation described above.
[0086] In another aspect, the invention provides a method for identifying a subject infected with the inventive hEbola virus described above, including: (a) obtaining total RNA
from a biological sample obtained from the subject; (b) reverse transcribing the total RNA to obtain cDNA; and (c) amplifying the cDNA using a set of primers derived from a nucleotide sequence of the inventive 5 hEbola virus described above.
[0087] In one embodiment of the present invention, the set of primers are derived from the nucleotide sequence of the genome of the hEbola virus of Deposit Accession No.
200706291. In another, the set of primers are derived from the nucleotide sequence of SEQ ID
NOs: 1 or 10 or any of the inventive nucleotide sequences as described above, or a complement thereof.
10 [0088] The invention further relates to the use of the sequence information of the isolated virus for diagnostic and therapeutic methods. In a specific embodiment, the invention provides nucleic acid molecules which are suitable for use as primers consisting of or including the nucleotide sequence of SEQ ID NOs: 1 or 10, or a complement thereof, or at least a portion of the nucleotide sequence thereof. In another specific embodiment, the invention provides nucleic acid molecules 15 which are suitable for hybridization to the inventive hEbola nucleic acid;
including, but not limited to PCR primers, Reverse Transcriptase primers, probes for Southern analysis or other nucleic acid hybridization analysis for the detection of hEbola nucleic acids, e.g., consisting of or including the nucleotide sequence of SEQ ID NOs: 1, 10 a combination thereof, a complement thereof, or a portion thereof. The invention further encompasses chimeric or recombinant viruses encoded in 20 whole or in part by the nucleotide sequences.
[0089] In another aspect, the present invention provides methods for screening antiviral agents that inhibit the infectivity or replication of hEbola virus or variants thereof.
[0090] The invention further provides methods of preparing recombinant or chimeric forms of hEbola.
[0091] In another aspect, the invention provides vaccine preparations including the hEbola virus, including recombinant and chimeric forms of the virus, or subunits of the virus. The present invention encompasses recombinant or chimeric viruses encoded by viral vectors derived from the genome of the inventive hEbola virus described herein or natural variants thereof. In a specific embodiment, a recombinant virus is one derived from the hEbola virus of Deposit Accession No.
200706291. It is recognized that natural variants of the inventive hEbola viruses described herein comprise one or more mutations, including, but not limited to, point mutations, rearrangements, insertions, deletions etc., to the genomic sequence. It is recognized that the mutations may or may not result in a phenotypic change.
[0092] In another specific embodiment, a chimeric virus of the invention is a recombinant hEbola EboBun or EboIC virus which further comprises a heterologous nucleotide sequence. In accordance with the invention, a chimeric virus may be encoded by a nucleotide sequence in which heterologous nucleotide sequences have been added to the genome or in which endogenous or native nucleotide sequences have been replaced with heterologous nucleotide sequences.
[0093] According to the present invention, the chimeric viruses are encoded by the viral vectors of the invention which further comprise a heterologous nucleotide sequence. In accordance with the present invention a chimeric virus is encoded by a viral vector that may or may not include nucleic acids that are non-native to the viral genome. In accordance with the invention a chimeric virus is encoded by a viral vector to which heterologous nucleotide sequences have been added, inserted or substituted for native or non-native sequences. In accordance with the present invention, the chimeric virus may be encoded by nucleotide sequences derived from different species or variants of hEbola virus. In particular, the chimeric virus is encoded by nucleotide sequences that encode antigenic polypeptides derived from different species or variants of hEbola virus.
[0094] A chimeric virus may be of particular use for the generation of recombinant vaccines protecting against two or more viruses (Tao et al., J. Virol. 72, 2955-2961;
Durbin et al., 2000, J.
Virol. 74, 6821-6831; Skiadopoulos et al., 1998, J. Virol. 72, 1762-1768 (1998); Teng et al., 2000, J.
Virol. 74, 9317-9321). For example, it can be envisaged that a virus vector derived from the hEbola virus expressing one or more proteins of variants of hEbola virus including hEbola EboBun, or vice versa, will protect a subject vaccinated with such vector against infections by both the native hEbola and the variant. Attenuated and replication-defective viruses may be of use for vaccination purposes with live vaccines as has been suggested for other viruses. (See, for example, PCT WO 02/057302, at pp. 6 and 23; and United States Patent Application Publication 2008/0069838 incorporated by reference herein).
[0095] In accordance with the present invention the heterologous sequence to be incorporated into the viral vectors encoding the recombinant or chimeric viruses of the invention include sequences obtained or derived from different species or variants of hEbola.
[0096] In certain embodiments, the chimeric or recombinant viruses of the invention are encoded by viral vectors derived from viral genomes wherein one or more sequences, intergenic regions, termini sequences, or portions or entire ORF have been substituted with a heterologous or non-native sequence. In certain embodiments of the invention, the chimeric viruses of the invention are encoded by viral vectors derived from viral genomes wherein one or more heterologous sequences have been inserted or added to the vector.
[0097] The selection of the viral vector may depend on the species of the subject that is to be treated or protected from a viral infection. If the subject is human, then an attenuated hEbola virus can be used to provide the antigenic sequences.
[0098] In accordance with the present invention, the viral vectors can be engineered to provide antigenic sequences which confer protection against infection by the inventive hEbola and natural variants thereof. The viral vectors may be engineered to provide one, two, three or more antigenic sequences. In accordance with the present invention the antigenic sequences may be derived from the same virus, from different species or variants of the same type of virus, or from different viruses.
[0099] The expression products and/or recombinant or chimeric virions obtained in accordance with the invention may advantageously be utilized in vaccine formulations. The expression products and chimeric virions of the present invention may be engineered to create vaccines against a broad range of pathogens, including viral and bacterial antigens, tumor antigens, allergen antigens, and auto antigens involved in autoimmune disorders. One way to achieve this goal involves modifying existing hEbola genes to contain foreign sequences in their respective external domains. Where the heterologous sequences are epitopes or antigens of pathogens, these chimeric viruses may be used to induce a protective immune response against the disease agent from which these determinants are derived. In particular, the chimeric virions of the present invention may be engineered to create vaccines for the protection of a subject from infections with hEbola virus and variants thereof.
[00100] Thus, the present invention further relates to the use of viral vectors and recombinant or chimeric viruses to formulate vaccines against a broad range of viruses and/or antigens. The present invention also encompasses recombinant viruses including a viral vector derived from the hEbola or variants thereof which contains sequences which result in a virus having a phenotype more suitable for use in vaccine formulations, e.g., attenuated phenotype or enhanced antigenicity. The mutations and modifications can be in coding regions, in intergenic regions and in the leader and trailer sequences of the virus.
[00101] The invention provides a host cell including a nucleic acid or a vector according to the invention. Plasmid or viral vectors containing the polymerase components of hEbola virus are generated in prokaryotic cells for the expression of the components in relevant cell types (bacteria, insect cells, eukaryotic cells). Plasmid or viral vectors containing full-length or partial copies of the hEbola genome will be generated in prokaryotic cells for the expression of viral nucleic acids in vitro or in vivo. The latter vectors optionally contain other viral sequences for the generation of chimeric viruses or chimeric virus proteins, optionally lack parts of the viral genome for the generation of replication defective virus, and optionally contain mutations, deletions or insertions for the generation of attenuated viruses. In addition, the present invention provides a host cell infected with hEbola virus of Deposit Accession No. 200706291, [00102] Infectious copies of West African hEbola (being wild type, attenuated, replication-defective or chimeric) are optionally produced upon co-expression of the polymerase components according to the state-of-the-art technologies described above.
[0100] In addition, eukaryotic cells, transiently or stably expressing one or more full-length or partial hEbola proteins are optionally used. Such cells are preferably made by transfection (proteins or nucleic acid vectors), infection (viral vectors) or transduction (viral vectors) and are useful for complementation of mentioned wild type, attenuated, replication-defective or chimeric viruses.
[0101] The viral vectors and chimeric viruses of the present invention optionally modulate a subject's immune system by stimulating a humoral immune response, a cellular immune response or by stimulating tolerance to an antigen. As used herein, a subject means:
humans, primates, horses, cows, sheep, pigs, goats, dogs, cats, avian species and rodents.
Formulation of Vaccines and Antivirals [0102] In a preferred embodiment, the invention provides a proteinaceous molecule or hEbola virus specific viral protein or functional fragment thereof encoded by a nucleic acid according to the invention. Useful proteinaceous molecules are for example derived from any of the genes or genomic fragments derivable from the virus according to the invention, preferably the GP, L, NP, sGP, VP24, VP30, VP35, and VP 40 proteins described herein. Such molecules, or antigenic fragments thereof, as provided herein, are for example useful in diagnostic methods or kits and in pharmaceutical compositions such as subunit vaccines. Particularly useful are polypeptides encoded by the nucleotide sequence of SEQ ID NOs: 1 or 10; or antigenic fragments thereof for inclusion as antigen or subunit immunogen, but inactivated whole virus can also be used.
Particularly useful are also those proteinaceous substances that are encoded by recombinant nucleic acid fragments of the hEbola genome, of course preferred are those that are within the preferred bounds and metes of ORFs, in particular, for eliciting hEbola specific antibody or T cell responses, whether in vivo (e.g.
for protective or therapeutic purposes or for providing diagnostic antibodies) or in vitro (e.g. by phage display technology or another technique useful for generating synthetic antibodies).
[0103] It is recognized that numerous variants, analogues, or homologues of EboBun polypeptides are within the scope of the present invention including amino acid substitutions, alterations, modifications, or other amino acid changes that increase, decrease, or do not alter the function or immunogenic propensity of the inventive immunogen or vaccine.
Several post-translational modifications are similarly envisioned as within the scope of the present invention illustratively including incorporation of a non-naturally occurring amino acid(s), phosphorylation, glycosylation, sulfation, and addition of pendent groups such as biotynlation, fluorophores, lumiphores, radioactive groups, antigens, or other molecules.
[0104] Methods of expressing and purifying natural or recombinant peptides and proteins are well known in the art. Illustratively, peptides and proteins are recombinantly expressed in eukaryotic cells. Exemplary eukaryotic cells include yeast, HeLa cells, 293 cells, COS cells, Chinese hamster ovary cells (CHO), and many other cell types known in the art.
Both eukaryotic and prokaryotic expression systems and cells are available illustratively from Invitrogen Corp., Carlsbad, CA. It is appreciated that cell-free expression systems are similarly operable.
[0105] In a preferred embodiment an immunogenic polypeptide is a full length EboBun protein.
Preferably, an immunogen is a full length EboBun protein of SEQ ID NOs: 2-9 or 59, or EboIC SEQ
ID NOs: 11-19, or a fragment thereof as described herein. Preferably, an immunogen is has a minimum of 5 amino acids. As used herein an immunogen is preferably a polypeptide. In the context of an immunogenic polypeptide the terms immunogen, polypeptide, and antigen are used interchangeably.
[0106] Modifications and changes can be made in the structure of the inventive immunogens that are the subject of the application and still obtain a molecule having similar or improved characteristics as the wild-type sequence (e.g., a conservative amino acid substitution). For example, certain amino acids are optionally substituted for other amino acids in a sequence without appreciable loss of immunogenic activity. Because it is the interactive capacity and nature of a polypeptide that defines that polypeptide's biological functional activity, certain amino acid sequence substitutions can be made in a polypeptide sequence and nevertheless obtain a polypeptide with like or improved properties. Optionally, a polypeptide is used that has less or more immunogenic activity compared to the wild-type sequence.
[0107] In making such changes, the hydropathic index of amino acids is preferably considered.
The importance of the hydropathic amino acid index in conferring interactive biologic function on a polypeptide is generally understood in the art. It is known that certain amino acids can be substituted for other amino acids having a similar hydropathic index or score and still result in a 5 polypeptide with similar biological activity. Each amino acid has been assigned a hydropathic index on the basis of its hydrophobicity and charge characteristics. Those indices are: isoleucine (+4.5);
valine (+4.2); leucine (+3.8); phenylalanine (+2.8); cysteine/cysteine (+2.5);
methionine (+1.9);
alanine (+1.8); glycine (-0.4); threonine (-0.7); serine (-0.8); tryptophan (-0.9); tyrosine (-1.3);
proline (-1.6); histidine (-3.2); glutamate (-3.5); glutamine (-3.5);
aspartate (-3.5); asparagine (-3.5);
10 lysine (-3.9); and arginine (-4.5).
[0108] It is believed that the relative hydropathic character of the amino acid determines the secondary structure of the resultant polypeptide, which in turn defines the interaction of the polypeptide with other molecules, such as enzymes, substrates, receptors, antibodies, antigens, and the like. It is known in the art that an amino acid can be substituted by another amino acid having a 15 similar hydropathic index and still obtain a functionally equivalent immunogen. In such changes, the substitution of amino acids whose hydropathic indices are within 2 is preferred, those within 1 are particularly preferred, and those within 0.5 are even more particularly preferred.
[0109] As outlined above, amino acid substitutions are generally based on the relative similarity of the amino acid side-chain substituents, for example, their hydrophobicity, hydrophilicity, charge, 20 size, and the like. Exemplary substitutions that take various of the foregoing characteristics into consideration are well known to those of skill in the art and include (original residue: exemplary substitution): (Ala: Gly, Ser), (Arg: Lys), (Asn: Gln, His), (Asp: Glu, Cys, Ser), (Gln: Asn), (Glu:
Asp), (Gly: Ala), (His: Asn, Gln), (Ile: Leu, Val), (Leu: Ile, Val), (Lys:
Arg), (Met: Leu, Tyr), (Ser:
Thr), (Thr: Ser), (Tip: Tyr), (Tyr: Trp, Phe), and (Val: Ile, Leu).
Embodiments of this disclosure 25 thus contemplate functional or biological equivalents of a polypeptide and immunogen as set forth above. In particular, embodiments of the polypeptides and immunogens optionally include variants having about 50%, 60%, 70%, 80%, 90%, and 95% sequence identity to the polypeptide of interest.
[0110] The invention provides vaccine formulations for the prevention and treatment of infections with hEbola virus. In certain embodiments, the vaccine of the invention comprises recombinant and chimeric viruses of the hEbola virus. In certain embodiments, the virus is attenuated.
[0111] In another embodiment of this aspect of the invention, inactivated vaccine formulations are prepared using conventional techniques to "kill" the chimeric viruses.
Inactivated vaccines are "dead" in the sense that their infectivity has been destroyed. Ideally, the infectivity of the virus is destroyed without affecting its immunogenicity. In order to prepare inactivated vaccines, the chimeric virus may be grown in cell culture or in the allantois of the chick embryo, purified by zonal ultracentrifugation, inactivated by formaldehyde or (3-propiolactone, and pooled. The resulting vaccine is usually inoculated intramuscularly or intranasally.
[0112] Inactivated viruses are optionally formulated with a suitable adjuvant in order to enhance the immunological response. Such adjuvants illustratively include but are not limited to mineral gels, e.g., aluminum hydroxide; surface active substances such as lysolecithin, pluronic polyols, polyanions; peptides; oil emulsions; and potentially useful human adjuvants such as BCG and Corynebacterium parvum.
[0113] In another aspect, the present invention also provides DNA vaccine formulations including a nucleic acid or fragment of the inventive hEbola virus, e.g., the virus having Accession No. 200706291, or nucleic acid molecules having the sequence of SEQ ID NOs: 1 or 10, or a fragment thereof. In another specific embodiment, the DNA vaccine formulations of the present invention comprise a nucleic acid or fragment thereof encoding the antibodies which immunospecifically bind hEbola viruses. In DNA vaccine formulations, a vaccine DNA comprises a viral vector, such as that derived from the hEbola virus, bacterial plasmid, or other expression vector, bearing an insert including a nucleic acid molecule of the present invention operably linked to one or more control elements, thereby allowing expression of the vaccinating proteins encoded by the nucleic acid molecule in a vaccinated subject. Such vectors can be prepared by recombinant DNA technology as recombinant or chimeric viral vectors carrying a nucleic acid molecule of the present invention.
[0114] A nucleic acid as used herein refers to single- or double-stranded molecules which are optionally DNA, including the nucleotide bases A, T, C and G, or RNA, including the bases A, U
(substitutes for T), C, and G. The nucleic acid may represent a coding strand or its complement.
Nucleic acids are optionally identical in sequence to the sequence which is naturally occurring or include alternative codons which encode the same amino acid as that which is found in the naturally occurring sequence. Furthermore, nucleic acids optionally include codons which represent conservative substitutions of amino acids as are well known in the art.
[0115] As used herein, the term "isolated nucleic acid" means a nucleic acid separated or substantially free from at least some of the other components of the naturally occurring organism, for example, the cell structural components commonly found associated with nucleic acids in a cellular environment and/or other nucleic acids. The isolation of nucleic acids is illustratively accomplished by techniques such as cell lysis followed by phenol plus chloroform extraction, followed by ethanol precipitation of the nucleic acids. The nucleic acids of this invention are illustratively isolated from cells according to methods well known in the art for isolating nucleic acids. Alternatively, the nucleic acids of the present invention are optionally synthesized according to standard protocols well described in the literature for synthesizing nucleic acids. Modifications to the nucleic acids of the invention are also contemplated, provided that the essential structure and function of the peptide or polypeptide encoded by the nucleic acid are maintained.
[0116] The nucleic acid encoding the peptide or polypeptide of this invention is optionally part of a recombinant nucleic acid construct comprising any combination of restriction sites and/or functional elements as are well known in the art which facilitate molecular cloning and other recombinant DNA manipulations. Thus, the present invention further provides a recombinant nucleic acid construct including a nucleic acid encoding a polypeptide of this invention.
[0117] Generally, it may be more convenient to employ as the recombinant polynucleotide a cDNA version of the polynucleotide. It is believed that the use of a cDNA
version will provide advantages in that the size of the gene will generally be much smaller and more readily employed to transfect the targeted cell than will a genomic gene, which will typically be up to an order of magnitude larger than the cDNA gene. However, the inventor does not exclude the possibility of employing a genomic version of a particular gene where desired.
[0118] As used herein, the terms "engineered" and "recombinant" cells are synonymous with "host" cells and are intended to refer to a cell into which an exogenous DNA
segment or gene, such as a cDNA or gene has been introduced. Therefore, engineered cells are distinguishable from naturally occurring cells which do not contain a recombinantly introduced exogenous DNA segment or gene. A host cell is optionally a naturally occurring cell that is transformed with an exogenous DNA segment or gene or a cell that is not modified. A host cell preferably does not possess a naturally occurring gene encoding RSV G protein. Engineered cells are, thus, cells having a gene or genes introduced through the hand of man. Recombinant cells illustratively include those having an introduced cDNA or genomic DNA, and also include genes positioned adjacent to a promoter not naturally associated with the particular introduced gene.
[0119] To express a recombinant encoded polypeptide in accordance with the present invention one optionally prepares an expression vector that comprises a polynucleotide under the control of one or more promoters. To bring a coding sequence "under the control of' a promoter, one positions the 5' end of the translational initiation site of the reading frame generally between about 1 and 50 nucleotides "downstream" of (i.e., 3' of) the chosen promoter. The "upstream"
promoter stimulates transcription of the inserted DNA and promotes expression of the encoded recombinant protein.
This is the meaning of "recombinant expression" in the context used here.
[0120] Many standard techniques are available to construct expression vectors containing the appropriate nucleic acids and transcriptional/translational control sequences in order to achieve protein or peptide expression in a variety of host-expression systems. Cell types available for expression include, but are not limited to, bacteria, such as E. coli and B.
subtilis transformed with recombinant phage DNA, plasmid DNA or cosmid DNA expression vectors.
[0121] Certain examples of prokaryotic hosts illustratively include E. coli strain RR1, E. coli LE392, E. coli B, E. coli 1776 (ATCC No. 31537) as well as E. coli W3110 (F-, lambda-, prototrophic, ATCC No. 273325); bacilli such as Bacillus subtilis; and other enterobacteria such as Salmonella typhimurium, Serratia marcescens, and various Pseudomonas species.
[0122] In general, plasmid vectors containing replicon and control sequences that are derived from species compatible with the host cell are used in connection with these hosts. The vector ordinarily carries a replication site, as well as marking sequences that are capable of providing phenotypic selection in transformed cells. For example, E. coli is often transformed using pBR322, a plasmid derived from an E. coli species. Plasmid pBR322 contains genes for ampicillin and tetracycline resistance and thus provides easy means for identifying transformed cells. The pBR322 plasmid, or other microbial plasmid or phage may also contain, or be modified to contain, promoters that can be used by the microbial organism for expression of its own proteins.
[0123] In addition, phage vectors containing replicon and control sequences that are compatible with the host microorganism are optionally used as transforming vectors in connection with these hosts. For example, the phage lambda is optionally utilized in making a recombinant phage vector that can be used to transform host cells, such as E. coli LE392.
[0124] Further useful vectors include pIN vectors and pGEX vectors, for use in generating glutathione S-transferase (GST) soluble fusion proteins for later purification and separation or cleavage. Other suitable fusion proteins are those with (3-galactosidase, ubiquitin, or the like.
[0125] Promoters that are most commonly used in recombinant DNA construction include the (3-lactamase (penicillinase), lactose and tryptophan (trp) promoter systems.
While these are the most commonly used, other microbial promoters have been discovered and utilized, and details concerning their nucleotide sequences have been published, enabling those of skill in the art to ligate them functionally with plasmid vectors.
[0126] For expression in Saccharomyces, the plasmid YRp7, for example, is commonly used.
This plasmid contains the trpl gene, which provides a selection marker for a mutant strain of yeast lacking the ability to grow in tryptophan, for example ATCC No. 44076 or PEP4-1. The presence of the trpl lesion as a characteristic of the yeast host cell genome then provides an effective environment for detecting transformation by growth in the absence of tryptophan.
[0127] Suitable promoting sequences in yeast vectors illustratively include the promoters for 3-phosphoglycerate kinase or other glycolytic enzymes, such as enolase, glyceraldehyde-3-phosphate dehydrogenase, hexokinase, pyruvate decarboxylase, phosphofructokinase, glucose-6-phosphate isomerase, 3-phosphoglycerate mutase, pyruvate kinase, triosephosphate isomerase, phosphoglucose isomerase, and glucokinase. In constructing suitable expression plasmids, the termination sequences associated with these genes are also preferably ligated into the expression vector 3' of the sequence desired to be expressed to provide polyadenylation of the mRNA and termination.
[0128] Other suitable promoters, which have the additional advantage of transcription controlled by growth conditions, illustratively include the promoter region for alcohol dehydrogenase 2, isocytochrome C, acid phosphatase, degradative enzymes associated with nitrogen metabolism, and the aforementioned glyceraldehyde-3-phosphate dehydrogenase, and enzymes responsible for maltose and galactose utilization.
[0129] In addition to microorganisms, cultures of cells derived from multicellular organisms are also operable as hosts. In principle, any such cell culture is operable, whether from vertebrate or invertebrate culture. In addition to mammalian cells, these include insect cell systems infected with recombinant virus expression vectors (e.g., baculovirus); and plant cell systems infected with recombinant virus expression vectors (e.g., cauliflower mosaic virus, CaMV;
tobacco mosaic virus, TMV) or transformed with recombinant plasmid expression vectors (e.g., Ti plasmid) containing one or more coding sequences.
[0130] In a useful insect system, Autographica californica nuclear polyhedrosis virus (AcNPV) is used as a vector to express foreign genes. The virus grows in Spodoptera frugiperda cells. The isolated nucleic acid coding sequences are cloned into non-essential regions (for example the polyhedron gene) of the virus and placed under control of an AcNPV promoter (for example, the polyhedron promoter). Successful insertion of the coding sequences results in the inactivation of the polyhedron gene and production of non-occluded recombinant virus (i.e., virus lacking the proteinaceous coat coded for by the polyhedron gene). These recombinant viruses are then used to 5 infect Spodoptera frugiperda cells in which the inserted gene is expressed (e.g., U.S. Patent No.
4,215,051).
[0131] Examples of useful mammalian host cell lines include VERO and HeLa cells, Chinese hamster ovary (CHO) cell lines, W138, BHK, COS-7, 293, HepG2, NIH3T3, RIN and MDCK cell lines. In addition, a host cell is preferably chosen that modulates the expression of the inserted 10 sequences, or modifies and processes the gene product in the specific fashion desired. Such modifications (e.g., glycosylation) and processing (e.g., cleavage) of protein products may be important for the function of the encoded protein.
[0132] Different host cells have characteristic and specific mechanisms for the post-translational processing and modification of proteins. Appropriate cell lines or host systems are 15 preferably chosen to ensure the correct modification and processing of the foreign protein expressed.
Expression vectors for use in mammalian cells ordinarily include an origin of replication (as necessary), a promoter located in front of the gene to be expressed, along with any necessary ribosome binding sites, RNA splice sites, polyadenylation site, and transcriptional terminator sequences. The origin of replication is preferably provided either by construction of the vector to 20 include an exogenous origin, such as may be derived from SV40 or other viral (e.g., Polyoma, Adeno, VSV, BPV) source, or may be provided by the host cell chromosomal replication mechanism. If the vector is integrated into the host cell chromosome, the latter is often sufficient.
[0133] The promoters are optionally derived from the genome of mammalian cells (e.g., metallothionein promoter) or from mammalian viruses (e.g., the adenovirus late promoter; the 25 vaccinia virus 7.5K promoter). Further, it is also possible, and may be desirable, to utilize promoter or control sequences normally associated with the desired gene sequence, provided such control sequences are compatible with the host cell systems.
[0134] A number of viral based expression systems are operable herein, for example, commonly used promoters are derived from polyoma, Adenovirus 2, Adenovirus 5, cytomegalovirus and 30 Simian Virus 40 (SV40). The early and late promoters of SV40 virus are useful because both are obtained easily from the virus as a fragment which also contains the SV40 viral origin of replication.
Smaller or larger SV40 fragments are also operable, particularly when there is included the approximately 250 bp sequence extending from the HindIll site toward the Bgll site located in the viral origin of replication.
[0135] In cases where an adenovirus is used as an expression vector, the coding sequences are preferably ligated to an adenovirus transcription/translation control complex, e.g., the late promoter and tripartite leader sequence. This chimeric gene is then optionally inserted in the adenovirus genome by in vitro or in vivo recombination. Insertion in a non-essential region of the viral genome (e.g., region El or E3) will result in a recombinant virus that is viable and capable of expressing proteins in infected hosts.
[0136] Specific initiation signals may also be required for efficient translation of the claimed isolated nucleic acid coding sequences. These signals include the ATG
initiation codon and adjacent sequences. Exogenous translational control signals, including the ATG
initiation codon, may additionally need to be provided. One of ordinary skill in the art would readily be capable of determining this need and providing the necessary signals. It is well known that the initiation codon must be in-frame (or in-phase) with the reading frame of the desired coding sequence to ensure translation of the entire insert. These exogenous translational control signals and initiation codons are optionally of a variety of origins, both natural and synthetic. The efficiency of expression is optionally enhanced by the inclusion of appropriate transcription enhancer elements or transcription terminators.
[0137] In eukaryotic expression, one will also typically desire to incorporate into the transcriptional unit an appropriate polyadenylation site if one was not contained within the original cloned segment. Typically, the poly A addition site is placed about 30 to 2000 nucleotides "downstream" of the termination site of the protein at a position prior to transcription termination.
[0138] For long-term, high-yield production of recombinant proteins, stable expression is preferred. For example, cell lines that stably express constructs encoding proteins are engineered.
Rather than using expression vectors that contain viral origins of replication, host cells are preferably transformed with vectors controlled by appropriate expression control elements (e.g., promoter, enhancer, sequences, transcription terminators, polyadenylation sites, etc.), and a selectable marker.
Following the introduction of foreign DNA, engineered cells may be allowed to grow for 1-2 days in an enriched medium, and then are switched to a selective medium. The selectable marker in the recombinant plasmid confers resistance to the selection and allows cells to stably integrate the plasmid into their chromosomes and grow to form foci, which in turn can be cloned and expanded into cell lines.
[0139] A number of selection systems are illustratively used, including, but not limited, to the herpes simplex virus thymidine kinase, hypoxanthine-guanine phosphoribosyltransferase and adenine phosphoribosyltransferase genes, in tk-, hgprt- or aprt- cells, respectively. Also, antimetabolite resistance is optionally used as the basis of selection for dhfr, which confers resistance to methotrexate; gpt, which confers resistance to mycophenolic acid; neo, which confers resistance to the aminoglycoside G-418; and hygro, which confers resistance to hygromycin. It is appreciated that numerous other selection systems are known in the art that are similarly operable in the present invention.
[0140] The nucleic acids encoding the peptides and polypeptides of this invention are optionally administered as nucleic acid vaccines. For the purposes of vaccine delivery, a nucleic acid encoding a peptide or polypeptide of this invention is preferably in an expression vector that includes viral nucleic acid including, but not limited to, vaccinia virus, adenovirus, retrovirus and/or adeno-associated virus nucleic acid. The nucleic acid or vector of this invention is optoinally in a liposome or a delivery vehicle which can be taken up by a cell via receptor-mediated or other type of endocytosis. The nucleic acid vaccines of this invention are preferably in a pharmaceutically acceptable carrier or administered with an adjuvant. The nucleic acids encoding the peptides and polypeptides of this invention can also be administered to cells in vivo or ex vivo.
[0141] It is contemplated that the isolated nucleic acids of the disclosure are optionally "overexpressed", i.e., expressed in increased levels relative to its natural expression in cells of its indigenous organism, or even relative to the expression of other proteins in the recombinant host cell. Such overexpression is assessed by a variety of methods illustratively including radio-labeling and/or protein purification. However, simple and direct methods are preferred, for example, those involving SDS/PAGE and protein staining or immunoblotting, followed by quantitative analyses, such as densitometric scanning of the resultant gel or blot. A specific increase in the level of the recombinant protein or peptide in comparison to the level in natural in transfected cells is indicative of overexpression, as is a relative abundance of the specific protein in relation to the other proteins produced by the host cell and, e.g., visible on a gel.
[0142] Various heterologous vectors are described for DNA vaccinations against viral infections. For example, the vectors described in the following references, incorporated herein by reference, may be used to express hEbola sequences instead of the sequences of the viruses or other pathogens described; in particular, vectors described for hepatitis B virus (Michel, M. L. et al., 1995, DAN-mediated immunization to the hepatitis B surface antigen in mice: Aspects of the humoral response mimic hepatitis B viral infection in humans, Proc. Natl. Aca. Sci.
USA 92:5307-5311;
Davis, H. L. et al., 1993, DNA-based immunization induces continuous secretion of hepatitis B
surface antigen and high levels of circulating antibody, Human Molec. Genetics 2:1847-1851), HIV
virus (Wang, B. et al., 1993, Gene inoculation generates immune responses against human immunodeficiency virus type 1, Proc. Natl. Acad. Sci. USA 90:4156-4160; Lu, S.
et al., 1996, Simian immunodeficiency virus DNA vaccine trial in Macques, J. Virol. 70:3978-3991; Letvin, N.
L. et al., 1997, Potent, protective anti-HIV immune responses generated by bimodal HIV envelope DNA plus protein vaccination, Proc Natl Acad Sci USA. 94(17):9378-83), and influenza viruses (Robinson, H L et al., 1993, Protection against a lethal influenza virus challenge by immunization with a haemagglutinin-expressing plasmid DNA, Vaccine 11:957-960; Ulmer, J. B.
et al., Heterologous protection against influenza by injection of DNA encoding a viral protein, Science 259:1745-1749), as well as bacterial infections, such as tuberculosis (Tascon, R. E. et al., 1996, Vaccination against tuberculosis by DNA injection, Nature Med. 2:888-892;
Huygen, K. et al., 1996, Immunogenicity and protective efficacy of a tuberculosis DNA vaccine, Nature Med., 2:893-898), and parasitic infection, such as malaria (Sedegah, M., 1994, Protection against malaria by immunization with plasmid DNA encoding circumsporozoite protein, Proc. Natl.
Acad. Sci. USA
91:9866-9870; Doolan, D. L. et al., 1996, Circumventing genetic restriction of protection against malaria with multigene DNA immunization: CD8+T cell-interferon .delta., and nitric oxide-dependent immunity, J. Exper. Med., 1183:1739-1746).
[0143] Many methods are optionally used to introduce the vaccine formulations described above. These include, but are not limited to, oral, intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous, and intranasal routes. Alternatively, in a preferred embodiment the chimeric virus vaccine formulation is introduced via the natural route of infection of the pathogen for which the vaccine is designed. The DNA vaccines of the present invention are optionally administered in saline solutions by injections into muscle or skin using a syringe and needle (Wolff J. A. et al., 1990, Direct gene transfer into mouse muscle in vivo, Science 247:1465-1468; Raz, E., 1994, Intradermal gene immunization: The possible role of DNA uptake in the induction of cellular immunity to viruses, c. Natl. Acd. Sci. USA 91:9519-9523). Another way to administer DNA
vaccines operable herein is called the "gene gun" method, whereby microscopic gold beads coated with the DNA molecules of interest is fired into cells (Tang, D. et al., 1992, Genetic immunization is a simple method for eliciting an immune response, Nature 356:152-154). For general reviews of the methods for DNA vaccines, see Robinson, H. L., 1999, DNA vaccines: basic mechanism and immune responses (Review), Int. J. Mol. Med. 4(5):549-555; Barber, B., 1997, Introduction:
Emerging vaccine strategies, Seminars in Immunology 9(5):269-270; and Robinson, H. L. et al., 1997, DNA vaccines, Seminars in Immunology 9(5):271-283.
Attenuation of hEbola Virus or Variants Thereof [0144] The hEbola virus or variants thereof of the invention are optionally genetically engineered to exhibit an attenuated phenotype. In particular, the viruses of the invention exhibit an attenuated phenotype in a subject to which the virus is administered as a vaccine. Attenuation can be achieved by any method known to a skilled artisan. Without being bound by theory, the attenuated phenotype of the viruses of the invention is caused, e.g., by using a virus that naturally does not replicate well in an intended host species, for example, by reduced replication of the viral genome, by reduced ability of the virus to infect a host cell, or by reduced ability of the viral proteins to assemble to an infectious viral particle relative to the wild type species of the virus.
[0145] The attenuated phenotypes of hEbola virus or variants thereof are optionally tested by any method known to the artisan. A candidate virus, for example, is optionally tested for its ability to infect a host or for the rate of replication in a cell culture system. In certain embodiments, growth curves at different temperatures are used to test the attenuated phenotype of the virus. For example, an attenuated virus is able to grow at 35 C, but not at 39 C or 40 C. In certain embodiments, different cell lines are used to evaluate the attenuated phenotype of the virus. For example, an attenuated virus may only be able to grow in monkey cell lines but not the human cell lines, or the achievable virus titers in different cell lines are different for the attenuated virus. In certain embodiments, viral replication in the respiratory tract of a small animal model, including but not limited to, hamsters, cotton rats, mice and guinea pigs, is used to evaluate the attenuated phenotypes of the virus. In other embodiments, the immune response induced by the virus, including but not limited to, the antibody titers (e.g., assayed by plaque reduction neutralization assay or ELISA) is used to evaluate the attenuated phenotypes of the virus. In a specific embodiment, the plaque reduction neutralization assay or ELISA is carried out at a low dose. In certain embodiments, the ability of the hEbola virus to elicit pathological symptoms in an animal model is tested. A reduced ability of the virus to elicit pathological symptoms in an animal model system is indicative of its attenuated phenotype. In a specific embodiment, the candidate viruses are tested in a monkey model for nasal infection, indicated by mucus production.
[0146] The viruses of the invention are optionally attenuated such that one or more of the functional characteristics of the virus are impaired. In certain embodiments, attenuation is measured in comparison to the wild type species of the virus from which the attenuated virus is derived. In other embodiments, attenuation is determined by comparing the growth of an attenuated virus in 5 different host systems. Thus, for a non-limiting example, hEbola virus or a variant thereof is attenuated when grown in a human host if the growth of the hEbola or variant thereof in the human host is reduced compared to the non-attenuated hEbola or variant thereof.
[0147] In certain embodiments, the attenuated virus of the invention is capable of infecting a host, is capable of replicating in a host such that infectious viral particles are produced. In 10 comparison to the wild type species, however, the attenuated species grows to lower titers or grows more slowly. Any technique known to the skilled artisan can be used to determine the growth curve of the attenuated virus and compare it to the growth curve of the wild type virus.
[0148] In certain embodiments, the attenuated virus of the invention (e.g., a recombinant or chimeric hEbola) cannot replicate in human cells as well as the wild type virus (e.g., wild type 15 hEbola) does. However, the attenuated virus can replicate well in a cell line that lacks interferon functions, such as Vero cells.
[0149] In other embodiments, the attenuated virus of the invention is capable of infecting a host, of replicating in the host, and of causing proteins of the virus of the invention to be inserted into the cytoplasmic membrane, but the attenuated virus does not cause the host to produce new infectious 20 viral particles. In certain embodiments, the attenuated virus infects the host, replicates in the host, and causes viral proteins to be inserted in the cytoplasmic membrane of the host with the same efficiency as the wild type hEbola. In other embodiments, the ability of the attenuated virus to cause viral proteins to be inserted into the cytoplasmic membrane into the host cell is reduced compared to the wild type virus. In certain embodiments, the ability of the attenuated hEbola virus to replicate in 25 the host is reduced compared to the wild type virus. Any technique known to the skilled artisan can be used to determine whether a virus is capable of infecting a mammalian cell, of replicating within the host, and of causing viral proteins to be inserted into the cytoplasmic membrane of the host.
[0150] In certain embodiments, the attenuated virus of the invention is capable of infecting a host. In contrast to the wild type hEbola, however, the attenuated hEbola cannot be replicated in the 30 host. In a specific embodiment, the attenuated hEbola virus can infect a host and can cause the host to insert viral proteins in its cytoplasmic membranes, but the attenuated virus is incapable of being replicated in the host. Any method known to the skilled artisan can be used to test whether the attenuated hEbola has infected the host and has caused the host to insert viral proteins in its cytoplasmic membranes.
[0151] In certain embodiments, the ability of the attenuated virus to infect a host is reduced compared to the ability of the wild type virus to infect the same host. Any technique known to the skilled artisan can be used to determine whether a virus is capable of infecting a host.
[0152] In certain embodiments, mutations (e.g., missense mutations) are introduced into the genome of the virus, for example, into the sequence of SEQ ID NOs: 1 or 10, or to generate a virus with an attenuated phenotype. Mutations (e.g., missense mutations) can be introduced into the structural genes and/or regulatory genes of the hEbola. Mutations are optionally additions, substitutions, deletions, or combinations thereof. Such variant of hEbola can be screened for a predicted functionality, such as infectivity, replication ability, protein synthesis ability, assembling ability, as well as cytopathic effect in cell cultures. In a specific embodiment, the missense mutation is a cold-sensitive mutation. In another embodiment, the missense mutation is a heat-sensitive mutation. In another embodiment, the missense mutation prevents a normal processing or cleavage of the viral proteins.
[0153] In other embodiments, deletions are introduced into the genome of the hEbola virus, which result in the attenuation of the virus.
[0154] In certain embodiments, attenuation of the virus is achieved by replacing a gene of the wild type virus with a gene of a virus of a different species, of a different subgroup, or of a different variant. In another aspect, attenuation of the virus is achieved by replacing one or more specific domains of a protein of the wild type virus with domains derived from the corresponding protein of a virus of a different species. In certain other embodiments, attenuation of the virus is achieved by deleting one or more specific domains of a protein of the wild type virus.
[0155] When a live attenuated vaccine is used, its safety should also be considered. The vaccine preferably does not cause disease. Any techniques known in the art for improving vaccine safety are operable in the present invention. In addition to attenuation techniques, other techniques are optionally be used. One non-limiting example is to use a soluble heterologous gene that cannot be incorporated into the virion membrane. For example, a single copy of the soluble version of a viral transmembrane protein lacking the transmembrane and cytosolic domains thereof is used.
[0156] Various assays are optionally used to test the safety of a vaccine. For example, sucrose gradients and neutralization assays are used to test the safety. A sucrose gradient assay is optionally used to determine whether a heterologous protein is inserted in a virion. If the heterologous protein is inserted in the virion, the virion is preferably tested for its ability to cause symptoms in an appropriate animal model since the virus may have acquired new, possibly pathological, properties.
5.4 Adjuvants and Carrier Molecules [0157] hEbola-associated antigens are administered with one or more adjuvants.
In one embodiment, the hEbola-associated antigen is administered together with a mineral salt adjuvants or mineral salt gel adjuvant. Such mineral salt and mineral salt gel adjuvants include, but are not limited to, aluminum hydroxide (ALHYDROGEL, REHYDRAGEL), aluminum phosphate gel, aluminum hydroxyphosphate (ADJU-PHOS), and calcium phosphate.
[0158] In another embodiment, hEbola-associated antigen is administered with an immunostimulatory adjuvant. Such class of adjuvants include, but are not limited to, cytokines (e.g., interleukin-2, interleukin-7, interleukin-12, granulocyte-macrophage colony stimulating factor (GM-CSF), interferon-y interleukin-1(3 (IL-1 (3), and IL-1 0 peptide or Sclavo Peptide), cytokine-containing liposomes, triterpenoid glycosides or saponins (e.g., QuilA and QS-21, also sold under the trademark STIMULON, ISCOPREP), Muramyl Dipeptide (MDP) derivatives, such as N-acetyl-muramyl-L-threonyl-D-isoglutamine (Threonyl-MDP, sold under the trademark TERMURTIDE), GMDP, N-acetyl-nor-muramyl-L-alanyl-D-isoglutamine, N-acetylmuramyl-L-alanyl-D-isoglutaminyl-L-alanine-2-(1'-2'-dipalmitoyl-s- n-glycero-3-hydroxy phosphoryloxy)-ethylamine, muramyl tripeptide phosphatidylethanolamine (MTP-PE), unmethylated CpG
dinucleotides and oligonucleotides, such as bacterial DNA and fragments thereof, LPS, monophosphoryl Lipid A (3D-MLA sold under the trademark MPL), and polyphosphazenes.
[0159] In another embodiment, the adjuvant used is a particular adjuvant, including, but not limited to, emulsions, e.g., Freund's Complete Adjuvant, Freund's Incomplete Adjuvant, squalene or squalane oil-in-water adjuvant formulations, such as SAF and MF59, e.g., prepared with block-copolymers, such as L-121 (polyoxypropylene/polyoxyetheylene) sold under the trademark PLURONIC L-121, Liposomes, Virosomes, cochleates, and immune stimulating complex, which is sold under the trademark ISCOM.
[0160] In another embodiment, a microparticular adjuvant is used.
Microparticular adjuvants include, but are not limited to, biodegradable and biocompatible polyesters, homo- and copolymers of lactic acid (PLA) and glycolic acid (PGA), poly(lactide-co-glycolides) (PLGA) microparticles, polymers that self-associate into particulates (poloxamer particles), soluble polymers (polyphosphazenes), and virus-like particles (VLPs) such as recombinant protein particulates, e.g., hepatitis B surface antigen (HbsAg).
[0161] Yet another class of adjuvants that are optionally used include mucosal adjuvants, including but not limited to heat-labile enterotoxin from Escherichia coli (LT), cholera holotoxin (CT) and cholera Toxin B Subunit (CTB) from Vibrio cholerae, mutant toxins (e.g., LTK63 and LTR72), microparticles, and polymerized liposomes.
[0162] In other embodiments, any of the above classes of adjuvants are optionally used in combination with each other or with other adjuvants. For example, non-limiting examples of combination adjuvant preparations used to administer the hEbola-associated antigens of the invention include liposomes containing immunostimulatory protein, cytokines, T-cell and/or B-cell peptides, or microbes with or without entrapped IL-2 or microparticles containing enterotoxin.
Other adjuvants known in the art are also included within the scope of the invention (see Vaccine Design: The Subunit and Adjuvant Approach, Chap. 7, Michael F. Powell and Mark J. Newman (eds.), Plenum Press, New York, 1995, which is incorporated herein in its entirety).
[0163] The effectiveness of an adjuvant is illustratively determined by measuring the induction of antibodies directed against an immunogenic polypeptide containing a hEbola polypeptide epitope, the antibodies resulting from administration of this polypeptide in vaccines which are also comprised of the various adjuvants.
[0164] The polypeptides are optionally formulated into the vaccine as neutral or salt forms.
Pharmaceutically acceptable salts include the acid additional salts (formed with free amino groups of the peptide) and which are formed with inorganic acids, such as, for example, hydrochloric or phosphoric acids, or organic acids such as acetic, oxalic, tartaric, maleic, and the like. Salts formed with free carboxyl groups are optionally derived from inorganic bases, such as, for example, sodium potassium, ammonium, calcium, or ferric hydroxides, and such organic bases as isopropylamine, trimethylamine, 2-ethylamino ethanol, histidine, procaine and the like.
[0165] The vaccines of the invention are preferably multivalent or univalent.
Multivalent vaccines are made from recombinant viruses that direct the expression of more than one antigen.
[0166] Many methods are operable herein to introduce the vaccine formulations of the invention; these include but are not limited to oral, intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous, intranasal routes, and via scarification (scratching through the top layers of skin, e.g., using a bifurcated needle).
[0167] The patient to which the vaccine is administered is preferably a mammal, most preferably a human, but is also optionally a non-human animal including but not limited to lower primates, cows, horses, sheep, pigs, fowl (e.g., chickens), goats, cats, dogs, hamsters, mice and rats.
Preparation of Antibodies [0168] Antibodies that specifically recognize a polypeptide of the invention, such as, but not limited to, polypeptides including the sequence of SEQ ID NOs: 2-9, 59, or 11-19 and other polypeptides as described herein, or hEbola epitope or antigen-binding fragments thereof are used in a preferred embodiment for detecting, screening, and isolating the polypeptide of the invention or fragments thereof, or similar sequences that might encode similar enzymes from the other organisms. For example, in one specific embodiment, an antibody which immunospecifically binds hEbola epitope, or a fragment thereof, is used for various in vitro detection assays, including enzyme-linked immunosorbent assays (ELISA), radioimmunoassays, western blot, etc., for the detection of a polypeptide of the invention or, preferably, hEbola, in samples, for example, a biological material, including cells, cell culture media (e.g., bacterial cell culture media, mammalian cell culture media, insect cell culture media, yeast cell culture media, etc.), blood, plasma, serum, tissues, sputum, naseopharyngeal aspirates, etc.
[0169] Antibodies specific for a polypeptide of the invention or any epitope of hEbola are optionally generated by any suitable method known in the art. Polyclonal antibodies to an antigen of interest, for example, the hEbola virus from Deposit Accession No. 200706291, or including a nucleotide sequence of SEQ ID NOs: 1 or 10, are optionally produced by various procedures well known in the art. For example, an antigen is optionally administered to various host animals including, but not limited to, rabbits, mice, rats, etc., to induce the production of antisera containing polyclonal antibodies specific for the antigen. Various adjuvants are optionally used to increase the immunological response, depending on the host species, and include but are not limited to, Freund's (complete and incomplete) adjuvant, mineral gels such as aluminum hydroxide, surface active substances such as lysolecithin, pluronic polyols, polyanions, peptides, oil emulsions, keyhole limpet hemocyanins, dinitrophenol, and potentially useful adjuvants for humans such as BCG (Bacille Calmette-Guerin) and Corynebacterium parvum. Such adjuvants are also well known in the art.
[0170] Monoclonal antibodies are optionally prepared using a wide variety of techniques known in the art including the use of hybridoma, recombinant, and phage display technologies, or a combination thereof. In one example, monoclonal antibodies are produced using hybridoma techniques including those known in the art and taught, for example, in Harlow et al., Antibodies: A
Laboratory Manual (Cold Spring Harbor Laboratory Press, 2nd ed. 1988);
Hammerling, et al., in:
Monoclonal Antibodies and T-Cell Hybridomas, pp. 563-681 (Elsevier, N.Y., 1981) (both of which are incorporated by reference in their entireties). The term "monoclonal antibody" as used herein is not limited to antibodies produced through hybridoma technology. The term "monoclonal antibody"
refers to an antibody that is derived from a single clone, including any eukaryotic, prokaryotic, or phage clone, and not the method by which it is produced.
5 [0171] Methods for producing and screening for specific antibodies using hybridoma technology are routine and well known in the art. In a non-limiting example, mice are immunized with an antigen of interest or a cell expressing such an antigen. Once an immune response is detected, e.g., antibodies specific for the antigen are detected in the mouse serum, the mouse spleen is harvested and splenocytes isolated. The splenocytes are then fused by well known techniques to 10 any suitable myeloma cells. Hybridomas are selected and cloned by limiting dilution. The hybridoma clones are then assayed by methods known in the art for cells that secrete antibodies capable of binding the antigen. Ascites fluid, which generally contains high levels of antibodies, is optionally generated by inoculating mice intraperitoneally with positive hybridoma clones.
[0172] Antibody fragments which recognize specific epitopes are optionally generated by 15 known techniques. For example, Fab and F(ab')2 fragments are illustratively produced by proteolytic cleavage of immunoglobulin molecules, using enzymes such as papain (to produce Fab fragments) or pepsin (to produce F(ab')2 fragments). F(ab')2 fragments preferably contain the complete light chain, and the variable region, the CH1 region and the hinge region of the heavy chain.
[0173] The antibodies of the invention or fragments thereof are optionally produced by any 20 method known in the art for the synthesis of antibodies, in particular, by chemical synthesis or preferably, by recombinant expression techniques.
[0174] The nucleotide sequence encoding an antibody is obtained from any information available to those skilled in the art (i.e., from Genbank, the literature, or by routine cloning and sequence analysis). If a clone containing a nucleic acid encoding a particular antibody or an epitope-25 binding fragment thereof is not available, but the sequence of the antibody molecule or epitope-binding fragment thereof is known, a nucleic acid encoding the immunoglobulin may be chemically synthesized or obtained from a suitable source (e.g., an antibody cDNA
library, or a cDNA library generated from, or nucleic acid, preferably poly A+RNA, isolated from any tissue or cells expressing the antibody, such as hybridoma cells selected to express an antibody) by PCR
amplification using 30 synthetic primers hybridizable to the 3' and 5' ends of the sequence or by cloning using an oligonucleotide probe specific for the particular gene sequence to identify, e.g., a cDNA clone from a cDNA library that encodes the antibody. Amplified nucleic acids generated by PCR are optionally then cloned into replicable cloning vectors using any method known in the art.
[0175] Once the nucleotide sequence of the antibody is determined, the nucleotide sequence of the antibody is optionally manipulated using methods well known in the art for the manipulation of nucleotide sequences, e.g., recombinant DNA techniques, site directed mutagenesis, PCR, etc. (see, for example, the techniques described in Sambrook et al., supra;, and Ausubel et al., eds., 1998, Current Protocols in Molecular Biology, John Wiley & Sons, NY, which are both incorporated by reference herein in their entireties), to generate antibodies having a different amino acid sequence by, for example, introducing amino acid substitutions, deletions, and/or insertions into the epitope-binding domain regions of the antibodies or any portion of antibodies which may enhance or reduce biological activities of the antibodies.
[0176] Recombinant expression of an antibody requires construction of an expression vector containing a nucleotide sequence that encodes the antibody. Once a nucleotide sequence encoding an antibody molecule or a heavy or light chain of an antibody, or portion thereof has been obtained, the vector for the production of the antibody molecule is optionally produced by recombinant DNA
technology using techniques known in the art as discussed in the previous sections. Methods which are known to those skilled in the art are optionally used to construct expression vectors containing antibody coding sequences and appropriate transcriptional and translational control signals. These methods include, for example, in vitro recombinant DNA techniques, synthetic techniques, and in vivo genetic recombination. The nucleotide sequence encoding the heavy-chain variable region, light-chain variable region, both the heavy-chain and light-chain variable regions, an epitope-binding fragment of the heavy- and/or light-chain variable region, or one or more complementarity determining regions (CDRs) of an antibody are optionally cloned into such a vector for expression.
Thus, prepared expression vector is optionally then introduced into appropriate host cells for the expression of the antibody. Accordingly, the invention includes host cells containing a polynucleotide encoding an antibody specific for the polypeptides of the invention or fragments thereof.
[0177] The host cell is optionally co-transfected with two expression vectors of the invention, the first vector encoding a heavy chain derived polypeptide and the second vector encoding a light chain derived polypeptide. The two vectors illustratively contain identical selectable markers which enable equal expression of heavy and light chain polypeptides or different selectable markers to ensure maintenance of both plasmids. Alternatively, a single vector is optionally used which encodes, and is capable of expressing, both heavy and light chain polypeptides. In such situations, the light chain should be placed before the heavy chain to avoid an excess of toxic free heavy chain (Proudfoot, Nature, 322:52, 1986; and Kohler, Proc. Natl. Acad. Sci. USA, 77:2 197, 1980). The coding sequences for the heavy and light chains optionally include cDNA or genomic DNA.
[0178] In another embodiment, antibodies are generated using various phage display methods known in the art. In phage display methods, functional antibody domains are displayed on the surface of phage particles which carry the polynucleotide sequences encoding them. In a particular embodiment, such phage is utilized to display antigen binding domains, such as Fab and Fv or disulfide-bond stabilized Fv, expressed from a repertoire or combinatorial antibody library (e.g., human or murine). Phage expressing an antigen binding domain that binds the antigen of interest is optionally selected or identified with antigen, e.g., using labeled antigen or antigen bound or captured to a solid surface or bead. Phages used in these methods are typically filamentous phage, including fd and M13. The antigen binding domains are expressed as a recombinantly fused protein to either the phage gene III or gene VIII protein. Examples of phage display methods that can be used to make the immunoglobulins, or fragments thereof, of the present invention include those disclosed in Brinkman et al., J. Immunol. Methods, 182:41-50, 1995; Ames et al., J. Immunol.
Methods, 184:177-186, 1995; Kettleborough et al., Eur. J. Immunol., 24:952-958, 1994; Persic et al., Gene, 187:9-18, 1997; Burton et al., Advances in Immunology, 57:191-280, 1994;
PCT application No. PCT/GB91/01134; PCT publications WO 90/02809; WO 91/10737; WO 92/01047; WO
92/18619; WO 93/11236; WO 95/15982; WO 95/20401; and U.S. Pat. Nos. 5,698,426;
5,223,409;
5,403,484; 5,580,717; 5,427,908; 5,750,753; 5,821,047; 5,571,698; 5,427,908;
5,516,637;
5,780,225; 5,658,727; 5,733,743 and 5,969,108; each of which is incorporated herein by reference in its entirety.
[0179] As described in the above references, after phage selection, the antibody coding regions from the phage is optionally isolated and used to generate whole antibodies, including human antibodies, or any other desired fragments, and expressed in any desired host, including mammalian cells, insect cells, plant cells, yeast, and bacteria, e.g., as described in detail below. For example, techniques to recombinantly produce Fab, Fab' and F(ab')2 fragments are optionally employed using methods known in the art such as those disclosed in PCT publication WO
92/22324; Mullinax et al., BioTechniques, 12(6):864-869, 1992; and Sawai et al., AJRI, 34:26-34, 1995;
and Better et al., Science, 240:1041-1043, 1988 (each of which is incorporated by reference in its entirety). Examples of techniques operable to produce single-chain Fvs and antibodies include those described in U.S.
Pat. Nos. 4,946,778 and 5,258,498; Huston et al., Methods in Enzymology, 203:46-88, 1991; Shu et al., PNAS, 90:7995-7999, 1993; and Skerra et al., Science, 240:1038-1040, 1988.
[0180] Once an antibody molecule of the invention has been produced by any methods described above, or otherwise known in the art, it is then optionally purified by any method known in the art for purification of an immunoglobulin molecule, for example, by chromatography (e.g., ion exchange, affinity, particularly by affinity for the specific antigen after Protein A or Protein G
purification, and sizing column chromatography), centrifligation, differential solubility, or by any other standard technique(s) for the purification of proteins. Further, the antibodies of the present invention or fragments thereof are optionally fused to heterologous polypeptide sequences described herein or otherwise known in the art to facilitate purification. Illustrative examples include 6xHis tag, FLAG tag, biotin, avidin, or other system.
[0181] For some uses, including in vivo use of antibodies in humans and in vitro detection assays, it is preferable to use chimeric, humanized, or human antibodies. A
chimeric antibody is a molecule in which different portions of the antibody are derived from different animal species, such as antibodies having a variable region derived from a murine monoclonal antibody and a constant region derived from a human immunoglobulin. Methods for producing chimeric antibodies are known in the art. See e.g., Morrison, Science, 229:1202, 1985; Oi et al., BioTechniques, 4:214 1986; Gillies et al., J. Immunol. Methods, 125:191-202, 1989; U.S. Pat. Nos.
5,807,715; 4,816,567;
and 4,816,397, which are incorporated herein by reference in their entireties.
Humanized antibodies are antibody molecules from non-human species that bind the desired antigen having one or more complementarity determining regions (CDRs) from the non-human species and framework regions from a human immunoglobulin molecule. Often, framework residues in the human framework regions will be substituted with the corresponding residue from the CDR donor antibody to alter, preferably improve, antigen binding. These framework substitutions are identified by methods well known in the art, e.g., by modeling of the interactions of the CDR and framework residues to identify framework residues important for antigen binding and sequence comparison to identify unusual framework residues at particular positions. See, e.g., Queen et al., U.S. Pat. No. 5,585,089;
Riechmann et al., Nature, 332:323, 1988, which are incorporated herein by reference in their entireties. Antibodies are humanized using a variety of techniques known in the art including, for example, CDR-grafting (EP 239,400; PCT publication WO 91/09967; U.S. Pat. Nos.
5,225,539;
5,530,101 and 5,585,089), veneering or resurfacing (EP 592,106; EP 519,596;
Padlan, Molecular Immunology, 28(4/5):489-498, 1991; Studnicka et al., Protein Engineering, 7(6):805-814, 1994;
Roguska et al., Proc Natl. Acad. Sci. USA, 91:969-973, 1994), and chain shuffling (U.S. Pat. No.
5,565,332), all of which are hereby incorporated by reference in their entireties.
[0182] Completely human antibodies are particularly desirable for therapeutic treatment of human patients. Human antibodies are made by a variety of methods known in the art illustratively including phage display methods described above using antibody libraries derived from human immunoglobulin sequences. See U.S. Pat. Nos. 4,444,887 and 4,716,111; and PCT
publications WO
98/46645; WO 98/50433; WO 98/24893; WO 98/16654; WO 96/34096; WO 96/33735; and WO
91/10741, each of which is incorporated herein by reference in its entirety.
[0183] Human antibodies are also illustratively produced using transgenic mice which are incapable of expressing functional endogenous immunoglobulins, but which can express human immunoglobulin genes. For an overview of this technology for producing human antibodies, see Lonberg and Huszar, Int. Rev. Immunol., 13:65-93, 1995. For a detailed discussion of this technology for producing human antibodies and human monoclonal antibodies and protocols for producing such antibodies, see, e.g., PCT publications WO 98/24893; WO
92/01047; WO 96/34096;
WO 96/33735; European Patent No. 0 598 877; U.S. Pat. Nos. 5,413,923;
5,625,126; 5,633,425;
5,569,825; 5,661,016; 5,545,806; 5,814,318; 5,885,793; 5,916,771; and 5,939,598, which are incorporated by reference herein in their entireties. In addition, companies such as Abgenix, Inc.
(Fremont, Calif.), Medarex (NJ) and Genpharm (San Jose, Calif.) can be engaged to provide human antibodies directed against a selected antigen using technology similar to that described above.
[0184] Completely human antibodies which recognize a selected epitope are optionally generated using a technique referred to as "guided selection." In this approach a selected non-human monoclonal antibody, e.g., a mouse antibody, is used to guide the selection of a completely human antibody recognizing the same epitope. (Jespers et al., Bio/technology, 12:899-903, 1988).
[0185] Antibodies fused or conjugated to heterologous polypeptides are optionally used in in vitro immunoassays and in purification methods (e.g., affinity chromatography) known in the art.
See e.g., PCT publication No. WO 93/21232; EP 439,095; Naramura et al., Immunol. Lett., 39:91-99, 1994; U.S. Pat. No. 5,474,981; Gillies et al., PNAS, 89:1428-1432, 1992;
and Fell et al., J.
Immunol., 146:2446-2452, 1991, which are incorporated herein by reference in their entireties.
[0186] Antibodies may also be illustratively attached to solid supports, which are particularly useful for immunoassays or purification of the polypeptides of the invention or fragments, derivatives, analogs, or variants thereof, or similar molecules having the similar enzymatic activities as the polypeptide of the invention. Such solid supports include, but are not limited to, glass, cellulose, polyacrylamide, nylon, polystyrene, polyvinyl chloride or polypropylene.
Pharmaceutical Compositions and Kits 5 [0187] The present invention encompasses pharmaceutical compositions including antiviral agents of the present invention. In a specific embodiment, the antiviral agent is preferably an antibody which immunospecifically binds and neutralizes the hEbola virus or variants thereof, or any proteins derived therefrom. In another specific embodiment, the antiviral agent is a polypeptide or nucleic acid molecule of the invention. The pharmaceutical compositions have utility as an 10 antiviral prophylactic agent are illustratively administered to a subject where the subject has been exposed or is expected to be exposed to a virus.
[0188] Various delivery systems are known and operable to administer the pharmaceutical composition of the invention, illustratively, encapsulation in liposomes, microparticles, microcapsules, recombinant cells capable of expressing the mutant viruses, and receptor mediated 15 endocytosis (see, e.g., Wu and Wu, 1987, J. Biol. Chem. 262:4429 4432).
Methods of introduction include but are not limited to intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous, intranasal, epidural, and oral routes. The compounds may be administered by any convenient route, for example by infusion or bolus injection, by absorption through epithelial or mucocutaneous linings (e.g., oral mucosa, rectal and intestinal mucosa, etc.) and optionally administered together 20 with other biologically active agents. Administration is systemic or local.
In a preferred embodiment, it is desirable to introduce the pharmaceutical compositions of the invention into the lungs by any suitable route. Pulmonary administration can also be employed, e.g., by use of an inhaler or nebulizer, and formulation with an aerosolizing agent.
[0189] In a specific embodiment, it is desirable to administer the pharmaceutical compositions 25 of the invention locally to the area in need of treatment. This administration may be achieved by, for example, and not by way of limitation, local infusion during surgery, topical application, e.g., in conjunction with a wound dressing after surgery, by injection, by means of a catheter, by means of a suppository, by means of nasal spray, or by means of an implant, the implant being of a porous, non-porous, or gelatinous material, including membranes, such as sialastic membranes, or fibers. In one 30 embodiment, administration can be by direct injection at the site (or former site) infected tissues.
[0190] In another embodiment, the pharmaceutical composition is delivered in a vesicle, in particular a liposome (see Langer, 1990, Science 249:1527-1533; Treat et al., in Liposomes in the Therapy of Infectious Disease and Cancer, Lopez Berestein and Fidler (eds.), Liss, New York, pp.
353-365 (1989); Lopez-Berestein, ibid. , pp. 317-327; see generally ibid.).
[0191] In yet another embodiment, the pharmaceutical composition is delivered in a controlled release system. In one embodiment, a pump is used (see Langer, supra; Sefton, 1987, CRC Crit.
Ref. Biomed. Eng. 14:201; Buchwald et al., 1980, Surgery 88:507; and Saudek et al., 1989, N. Engl.
J. Med. 321:574). In another embodiment, polymeric materials are used (see Medical Applications of Controlled Release, Langer and Wise (eds.), CRC Pres., Boca Raton, Fla.
(1974); Controlled Drug Bioavailability, Drug Product Design and Performance, Smolen and Ball (eds.), Wiley, New York (1984); Ranger and Peppas, J. Macromol. Sci. Rev. Macromol. Chem. 23:61 (1983); see also Levy et al., 1985, Science 228:190; During et al., 1989, Ann. Neurol. 25:351;
Howard et al., 1989, J.
Neurosurg. 71:105). In yet another embodiment, a controlled release system is placed in proximity of the composition's target, i.e., the lung, thus, requiring only a fraction of the systemic dose (see, e.g., Goodson, in Medical Applications of Controlled Release, supra, vol. 2, pp. 115-138 (1984)).
[0192] Other controlled release systems are discussed in the review by Langer (Science 249:1527-1533 (1990)) the contents of which are incorporated herein by reference.
[0193] The pharmaceutical compositions of the present invention illustratively include a therapeutically effective amount of a live attenuated, inactivated or killed West African hEbola virus, or recombinant or chimeric hEbola virus, and a pharmaceutically acceptable carrier. In a specific embodiment, the term "pharmaceutically acceptable" means approved by a regulatory agency of the Federal or a state government or listed in the U.S. Pharmacopeia or other generally recognized pharmacopeia for use in animals, and more particularly in humans.
The term "carrier"
refers to a diluent, adjuvant, excipient, or vehicle with which the pharmaceutical composition is administered. Such pharmaceutical carriers are illustratively sterile liquids, such as water and oils, including those of petroleum, animal, vegetable or synthetic origin, such as peanut oil, soybean oil, mineral oil, sesame oil and the like. Water is a preferred carrier when the pharmaceutical composition is administered intravenously. Saline solutions and aqueous dextrose and glycerol solutions are optionally employed as liquid carriers, particularly for injectable solutions. Suitable pharmaceutical excipients include starch, glucose, lactose, sucrose, gelatin, malt, rice, flour, chalk, silica gel, sodium stearate, glycerol monostearate, talc, sodium chloride, dried skim milk, glycerol, propylene, glycol, water, ethanol and the like. The composition, if desired, also contains wetting or emulsifying agents, or pH buffering agents. These compositions optionally take the form of solutions, suspensions, emulsion, tablets, pills, capsules, powders, sustained release formulations and the like. The composition is optionally formulated as a suppository, with traditional binders and carriers such as triglycerides. Oral formulation illustratively includes standard carriers such as pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharine, cellulose, magnesium carbonate, etc. Examples of suitable pharmaceutical carriers are described in "Remington's Pharmaceutical Sciences" by E. W. Martin. The formulation should suit the mode of administration.
[0194] In a preferred embodiment, the composition is formulated in accordance with routine procedures as a pharmaceutical composition adapted for intravenous administration to human beings. Typically, compositions for intravenous administration are solutions in sterile isotonic aqueous buffer. The composition also includes an optional solubilizing agent and a local anesthetic such as lignocaine to ease pain at the site of the injection. Generally, the ingredients are supplied either separately or mixed together in unit dosage form, for example, as a dry lyophilized powder or water-free concentrate in a hermetically sealed container such as an ampoule or sachette indicating the quantity of active agent. Where the composition is to be administered by infusion, it can be dispensed with an infusion bottle containing sterile pharmaceutical grade water or saline. Where the composition is administered by injection, an ampoule of sterile water for injection or saline is optionally provided so that the ingredients may be mixed prior to administration.
[0195] The pharmaceutical compositions of the invention are illustratively formulated as neutral or salt forms. Pharmaceutically acceptable salts illustratively include those formed with free amino groups such as those derived from hydrochloric, phosphoric, acetic, oxalic, tartaric acids, etc., and those formed with free carboxyl groups such as those derived from sodium, potassium, ammonium, calcium, ferric hydroxides, isopropylamine, triethylamine, 2 ethylamino ethanol, histidine, procaine, etc.
[0196] The amount of the pharmaceutical composition of the invention which will be effective in the treatment of a particular disorder or condition will depend on the nature of the disorder or condition, and can be determined by standard clinical techniques. In addition, in vitro assays are optionally employed to help identify optimal dosage ranges. The precise dose to be employed in the formulation will also depend on the route of administration, and the seriousness of the disease or disorder, and should be decided according to the judgment of the practitioner and each patient's circumstances. However, suitable dosage ranges for intravenous administration are generally about 20 to 500 micrograms of active compound per kilogram body weight. Suitable dosage ranges for intranasal administration are generally about 0.01 pg/kg body weight to 1 mg/kg body weight.
Effective doses may be extrapolated from dose response curves derived from in vitro or animal model test systems.
[0197] Suppositories generally contain active ingredient in the range of 0.5%
to 10% by weight;
oral formulations preferably contain 10% to 95% active ingredient.
[0198] The invention also provides a pharmaceutical pack or kit including one or more containers filled with one or more of the ingredients of the pharmaceutical compositions of the invention. Optionally associated with such container(s) is a notice in the form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals or biological products, which notice reflects approval by the agency of manufacture, use or sale for human administration. In a preferred embodiment, the kit contains an antiviral agent of the invention, e.g., an antibody specific for the polypeptides encoded by a nucleotide sequence of SEQ ID NOs: 1 or 10, or as shown in SEQ ID NOs: 2-9, 59, or 11-19, or any hEbola epitope, or a polypeptide or protein of the present invention, or a nucleic acid molecule of the invention, alone or in combination with adjuvants, antivirals, antibiotics, analgesic, bronchodilators, or other pharmaceutically acceptable excipients.
[0199] The present invention further encompasses kits including a container containing a pharmaceutical composition of the present invention and instructions for use.
Detection Assays [0200] The present invention provides a method for detecting an antibody, which immunospecifically binds to the hEbola virus, in a biological sample, including for example blood, serum, plasma, saliva, urine, feces, etc., from a patient suffering from hEbola infection, and/or hemorrhagic fever. In a specific embodiment, the method including contacting the sample with the hEbola virus, for example, of Deposit Accession No. 200706291, or having a genomic nucleic acid sequence of SEQ ID NOs: 1 or 10, directly immobilized on a substrate and detecting the virus-bound antibody directly or indirectly by a labeled heterologous anti-isotype antibody. In another specific embodiment, the sample is contacted with a host cell which is infected by the hEbola virus, for example, of Deposit Accession No. 200706291, or having a genomic nucleic acid sequence of SEQ
ID NOs: 1 or 10, and the bound antibody is optionally detected by immunofluorescent assay.
[0201] An exemplary method for detecting the presence or absence of a polypeptide or nucleic acid of the invention in a biological sample involves obtaining a biological sample from various sources and contacting the sample with a compound or an agent capable of detecting an epitope or nucleic acid (e.g., mRNA, genomic DNA) of the hEbola virus such that the presence of the hEbola virus is detected in the sample. A preferred agent for detecting hEbola mRNA
or genomic RNA of the invention is a labeled nucleic acid probe capable of hybridizing to mRNA
or genomic RNA
encoding a polypeptide of the invention. The nucleic acid probe is, for example, a nucleic acid molecule including the nucleotide sequence of SEQ ID NOs: 1 or 10, a complement thereof, or a portion thereof, such as an oligonucleotide of at least 15, 20, 25, 30, 50, 100, 250, 500, 750, 1000 or more contiguous nucleotides in length and sufficient to specifically hybridize under stringent conditions to a hEbola mRNA or genomic RNA.
[0202] As used herein, the term "stringent conditions" describes conditions for hybridization and washing under which nucleotide sequences having at least 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, or 95% identity to each other typically remain hybridized to each other. Such hybridization conditions are described in, for example but not limited to, Current Protocols in Molecular Biology, John Wiley & Sons, N.Y. (1989), 6.3.1 6.3.6.;
Basic Methods in Molecular Biology, Elsevier Science Publishing Co., Inc., N.Y. (1986), pp.75 78, and 84 87; and Molecular Cloning, Cold Spring Harbor Laboratory, N.Y. (1982), pp.387 389, and are well known to those skilled in the art. A preferred, non-limiting example of stringent hybridization conditions is hybridization in 6x sodium chloride/sodium citrate (SSC), 0.5% SDS at about 68 C followed by one or more washes in 2xSSC, 0.5% SDS at room temperature. Another preferred, non-limiting example of stringent hybridization conditions is hybridization in 6x SSC at about 45 C
followed by one or more washes in 0.2x SSC, 0.1% SDS at 50 to 65 C.
[0203] A nucleic acid probe, polynucleotide, oligonucleotide, or other nucleic acid is preferably purified. An "isolated" or "purified" nucleotide sequence is substantially free of cellular material or other contaminating proteins from the cell or tissue source from which the nucleotide is derived, or is substantially free of chemical precursors or other chemicals when chemically synthesized. The language "substantially free of cellular material" includes preparations of a nucleotide/oligonucleotide in which the nucleotide/oligonucleotide is separated from cellular components of the cells from which it is isolated or produced. Thus, a nucleotide/oligonucleotide that is substantially free of cellular material includes preparations of the nucleotide having less than about 30%, 20%, 10%, 5%, 2.5%, or 1%, (by dry weight) of contaminating material. When nucleotide/oligonucleotide is produced by chemical synthesis, it is preferably substantially free of chemical precursors or other chemicals, i.e., it is separated from chemical precursors or other chemicals which are involved in the synthesis of the protein. Accordingly, such preparations of the nucleotide/oligonucleotide have less than about 30%, 20%, 10%, or 5% (by dry weight) of chemical precursors or compounds other than the nucleotide/oligonucleotide of interest.
In a preferred embodiment of the present invention, the nucleotide/oligonucleotide is isolated or purified.
[0204] In another preferred specific embodiment, the presence of hEbola virus is detected in the 5 sample by a reverse transcription polymerase chain reaction (RT-PCR) using the primers that are constructed based on a partial nucleotide sequence of the genome of hEbola virus, for example, that of Deposit Accession No. 200706291, or having a genomic nucleic acid sequence of SEQ ID NOs: 1 or 10. In a non-limiting specific embodiment, preferred primers to be used in a RT-PCR method are the primers are described in detail herein.
10 [0205] In more preferred specific embodiment, the present invention provides a real-time quantitative PCR assay to detect the presence of hEbola virus in a biological sample by subjecting the cDNA obtained by reverse transcription of the extracted total RNA from the sample to PCR
reactions using the specific primers described in detail herein, and a fluorescence dye, such as SYBR Green I, which fluoresces when bound nonspecifically to double-stranded DNA. The 15 fluorescence signals from these reactions are captured at the end of extension steps as PCR product is generated over a range of the thermal cycles, thereby allowing the quantitative determination of the viral load in the sample based on an amplification plot.
[0206] A preferred agent for detecting hEbola is an antibody that specifically binds a polypeptide of the invention or any hEbola epitope, preferably an antibody with a detectable label.
20 Antibodies are illustratively polyclonal, or more preferably, monoclonal.
An intact antibody, or a fragment thereof (e.g., Fab or F(ab')2) is operable herein.
[0207] The term "labeled", with regard to the probe or antibody, is intended to encompass direct labeling of the probe or antibody by coupling (i.e., physically linking) a detectable substance to the probe or antibody, optionally via a linker, as well as indirect labeling of the probe or antibody by 25 reactivity with another reagent that is directly labeled. Examples of indirect labeling include detection of a primary antibody using a fluorescently labeled secondary antibody and end-labeling of a DNA probe with biotin such that it is detectable with fluorescently labeled streptavidin. The detection method of the invention is optionally used to detect mRNA, protein (or any epitope), or genomic RNA in a sample in vitro as well as in vivo. Exemplary in vitro techniques for detection of 30 mRNA include northern hybridizations, in situ hybridizations, RT-PCR, and RNase protection. In vitro techniques for detection of an epitope of hEbola illustratively include enzyme linked immunosorbent assays (ELISAs), western blots, immunoprecipitations and immunofluorescence. In vitro techniques for detection of genomic RNA include northern hybridizations, RT-PCT, and RNase protection. Furthermore, in vivo techniques for detection of hEbola include introducing into a subject organism a labeled antibody directed against the polypeptide. In one embodiment, the antibody is labeled with a radioactive marker whose presence and location in the subject organism is detected by standard imaging techniques, including autoradiography.
[0208] In a specific embodiment, the methods further involve obtaining a control sample from a control subject, contacting the control sample with a compound or agent capable of detecting hEbola, e.g., a polypeptide of the invention or mRNA or genomic RNA encoding a polypeptide of the invention, such that the presence of hEbola or the polypeptide or mRNA or genomic RNA
encoding the polypeptide is detected in the sample, and comparing the absence of hEbola or the polypeptide or mRNA or genomic RNA encoding the polypeptide in the control sample with the presence of hEbola, or the polypeptide or mRNA or genomic DNA encoding the polypeptide in the test sample.
[0209] The invention also encompasses kits for detecting the presence of hEbola or a polypeptide or nucleic acid of the invention in a test sample. The kit illustratively includes a labeled compound or agent capable of detecting hEbola or the polypeptide or a nucleic acid molecule encoding the polypeptide in a test sample and, in certain embodiments, a means for determining the amount of the polypeptide or mRNA in the sample (e.g., an antibody which binds the polypeptide or an oligonucleotide probe which binds to DNA or mRNA encoding the polypeptide).
Kits optionally include instructions for use.
[0210] For antibody-based kits, the kit illustratively includes: (1) a first antibody (e.g., attached to a solid support) which binds to a polypeptide of the invention or hEbola epitope; and, optionally, (2) a second, different antibody which binds to either the polypeptide or the first antibody and is preferably conjugated to a detectable agent.
[0211] For oligonucleotide-based kits, the kit illustratively includes: (1) an oligonucleotide, e.g., a detectably labeled oligonucleotide, which hybridizes to a nucleic acid sequence encoding a polypeptide of the invention or to a sequence within the hEbola genome; or (2) a pair of primers useful for amplifying a nucleic acid molecule containing an hEbola sequence.
The kit optionally includes a buffering agent, a preservative, or a protein stabilizing agent.
The kit optionally includes components necessary for detecting the detectable agent (e.g., an enzyme or a substrate). The kit optionally contains a control sample or a series of control samples which can be assayed and compared to the test sample contained. Each component of the kit is usually enclosed within an individual container and all of the various containers are within a single package along with instructions for use.
Screening Assays to Identify Antiviral Agents [0212] The invention provides methods for the identification of a compound that inhibits the ability of hEbola virus to infect a host or a host cell. In certain embodiments, the invention provides methods for the identification of a compound that reduces the ability of hEbola virus to replicate in a host or a host cell. Any technique well known to the skilled artisan is illustratively used to screen for a compound useful to abolish or reduce the ability of hEbola virus to infect a host and/or to replicate in a host or a host cell.
[0213] In certain embodiments, the invention provides methods for the identification of a compound that inhibits the ability of hEbola virus to replicate in a mammal or a mammalian cell.
More specifically, the invention provides methods for the identification of a compound that inhibits the ability of hEbola virus to infect a mammal or a mammalian cell. In certain embodiments, the invention provides methods for the identification of a compound that inhibits the ability of hEbola virus to replicate in a mammalian cell. In a specific embodiment, the mammalian cell is a human cell.
[0214] In another embodiment, a cell is contacted with a test compound and infected with the hEbola virus. In certain embodiments, a control culture is infected with the hEbola virus in the absence of a test compound. The cell is optionally contacted with a test compound before, concurrently with, or subsequent to the infection with the hEbola virus. In a specific embodiment, the cell is a mammalian cell. In an even more specific embodiment, the cell is a human cell. In certain embodiments, the cell is incubated with the test compound for at least 1 minute, at least 5 minutes, at least 15 minutes, at least 30 minutes, at least 1 hour, at least 2 hours, at least 5 hours, at least 12 hours, or at least 1 day. The titer of the virus is optionally measured at any time during the assay. In certain embodiments, a time course of viral growth in the culture is determined. If the viral growth is inhibited or reduced in the presence of the test compound, the test compound is identified as being effective in inhibiting or reducing the growth or infection of the hEbola virus. In a specific embodiment, the compound that inhibits or reduces the growth of the hEbola virus is tested for its ability to inhibit or reduce the growth rate of other viruses to test its specificity for the hEbola virus.
[0215] In one embodiment, a test compound is administered to a model animal and the model animal is infected with the hEbola virus. In certain embodiments, a control model animal is infected with the hEbola virus without the administration of a test compound. The test compound is optionally administered before, concurrently with, or subsequent to the infection with the hEbola virus. In a specific embodiment, the model animal is a mammal. In an even more specific embodiment, the model animal is, but is not limited to, a cotton rat, a mouse, or a monkey. The titer of the virus in the model animal is optionally measured at any time during the assay. In certain embodiments, a time course of viral growth in the culture is determined. If the viral growth is inhibited or reduced in the presence of the test compound, the test compound is identified as being effective in inhibiting or reducing the growth or infection of the hEbola virus. In a specific embodiment, the compound that inhibits or reduces the growth of the hEbola in the model animal is tested for its ability to inhibit or reduce the growth rate of other viruses to test its specificity for the hEbola virus.
[0216] According to the method of the invention, a human or an animal is optionally treated for for EboBun or EboIC, other viral infection or bacterial infection by administering an effective amount of an inventive therapeutic composition. Preferably, a vaccine is administered prophylactically. An "effective amount" is an amount that will induce an immune response in a subject. Illustratively, an effective amount of the compositions of this invention ranges from nanogram/kg to milligram/kg amounts for young children and adults. Equivalent dosages for lighter or heavier body weights can readily be determined. The dose should be adjusted to suit the individual to whom the composition is administered and will vary with age, weight and metabolism of the individual. The exact amount of the composition required will vary from subject to subject, depending on the species, age, weight and general condition of the subject, the particular peptide or polypeptide used, its mode of administration and the like. An appropriate amount can be determined by one of ordinary skill in the art using only routine experimentation given the teachings herein. One skilled in the art will realize that dosages are best optimized by the practicing physician or veterinarian and methods for determining dose amounts and regimens and preparing dosage forms are described, for example, in Remington's Pharmaceutical Sciences, (Martin, E. W., ed., latest edition), Mack Publishing Co., Easton, PA. Preferably, a single administration is operable to induce an immune response.
[0217] Methods involving conventional biological techniques are described herein. Such techniques are generally known in the art and are described in detail in methodology treatises such as Molecular Cloning: A Laboratory Manual, 2nd ed., vol. 1-3, ed. Sambrook et al., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1989; and Current Protocols in Molecular Biology, ed. Ausubel et al., Greene Publishing and Wiley-Interscience, New York, 1992 (with periodic updates). Immunological methods (e.g., preparation of antigen-specific antibodies, immunoprecipitation, and immunoblotting) are described, e.g., in Current Protocols in Immunology, ed. Coligan et al., John Wiley & Sons, New York, 1991; and Methods of Immunological Analysis, ed. Masseyeff et al., John Wiley & Sons, New York, 1992.
[0218] Embodiments of inventive compositions and methods are illustrated in the following detailed examples. These examples are provided for illustrative purposes and are not considered limitations on the scope of inventive compositions and methods.
EXAMPLES
Example 1:
Newly discovered Ebola virus associated with hemorrhagic fever outbreak in Bundibugyo, Uganda [0219] In late November 2007 HF cases were reported in the townships of Bundibugyo and Kikyo in Bundibugyo District, Western Uganda (Figure 1A). These samples were assayed as described by Towner, JS, et al., PLoS Pathog, 2008 November; 4(11): e1000212, the contents of which are incorporated herein by reference for methods, results, reagents, and all other aspects of the publication. A total of 29 blood samples were initially collected from suspect cases and showed evidence of acute ebolavirus infection in eight specimens using a broadly reactive ebolavirus antigen capture assay known to cross-react with the different ebolavirus species and an IgM capture assay based on Zaire ebolavirus reagents (Table 1). These specimens were negative when initially tested with highly sensitive real-time RT-PCR assays specific for all known Zaire and Sudan ebolaviruses and marburgviruses. However, further evidence of acute ebolavirus infection was obtained using a traditionally less sensitive (relative to the real-time RT-PCR assays) but more broadly reactive filovirus L gene-specific RT-PCR assay (1 specimen) (Table 1). Sequence analysis of the PCR
fragment (400 bp of the virus L gene) revealed the reason for the initial failure of the real-time RT-PCR assays, as the sequence was distinct from that of the 4 known species of ebolavirus, although distantly related to Cote d'Ivoire ebolavirus. In total, 9 of 29 specimens showed evidence of ebolavirus infection, and all tests were negative for marburgvirus (data not shown).
[0220] Approximately 70% of the virus genome was rapidly sequenced from total RNA
extracted from a patient serum (#200706291) using a newly established metagenomics pyrosequencing method (454 Life Sciences) which involves successive rounds of random DNA
amplification8. Using the newly derived draft sequence, a real-time RT-PCR
assay specific for the 5 NP gene of this virus was quickly developed and evaluated. The assay was shown to have excellent sensitivity (Table 1), finding positive all the initial six samples that tested positive by either virus antigen capture (five specimens) or virus isolation assays (four specimens).
The antigen-capture, IgM, IgG and newly designed real-time PCR assays were quickly transferred to the Uganda Virus Research Institute during the course of the outbreak to facilitate rapid identification and isolation of 10 Ebola cases in the affected area for efficient control of the outbreak. The outbreak continued through late December 2007, and resulted in 149 suspected cases and 37 deaths9.
[0221] Table 1. Ebolavirus diagnostic results of initial 29 specimens obtained from Bundibugyo District with numerical specimen numbers assigned. RT-PCR refers to results obtained from conventional PCR using the broadly reactive Filo A/B primers 13. Ag, IgM, and IgG refer to 15 results from ELISA-based assaysio' 11 with Zaire ebolavirus reagents while virus isolation refers to culture attempts on Vero E6 cells12. Q-RT-PCR refers to results obtained using the optimized Bundibugyo ebolavirus specific real-time RT-PCR assay with cycle threshold (Ct) values of positive (Pos) samples indicated in the far right column. * Specimen #200706291 is the clinical sample from which prototype isolate #811250 was obtained.
Table 1 ...............................................................................
...............................................................................
..................................... .
Sample No. RT-PCR Arc Virus Isolation Q RT PCR Ct ......... .........
200706288 neg neg neg neg neg neg 40 200706289 neg neg neg neg neg neg 40 200706290....:...... n eg......>....neg.......n eg.......n eg.
_............neg ............................ n eg.....................
40.......
200706291.......... Pos......:.... Pos.......n eg neg Pos ............................ Pos..................23.64...>
200706292..........neg..........!?.eg.......!?...9.......11e9.
_............!?.eg ............. ..............neg..............
.......40.......
!?.eg .............. .............. neg..............
200706293........... eg...... ....!?.eg.......!?...9........ neg.
.............
.......40.......
200706294 neg neg neg neg neg neg 40 .................................
..................................................................
....................................... ....................................
.....................
200706295 neg neg neg neg neg neg 40 200706296 neg neg Pos Pos neg neg 40 200706297 neg neg Pos Pos neg neg 40 200706298 neg Pos Pos Pos neg Pos 34.83 200706299 neg neg Pos Pos neg neg 40 200706300............neg........... neg..... ne..... neg..
.............neg............................ neg.....................40 200706301 ne. neg ne neg neg ne. 40 .................................;..............................
.............;.......... ..................
................................... ...................................
200706302 neg Pos Pos neg neg Pos 35.01 ................................:.....................:........................
....................
........................................................................:......
...............
200706303 neg neg neg neg neg neg 40 200706304 neg neg neg neg Pos Pos 38.18 200706305....:...... neg......>....neg.......neg.......ne9 _............neg ............................ neg.....................40.......>
200706306..........neg......>....neg.......neg......ne9 _............neg ............................neg.....................40.......
200706307..........neg......>....!?eg.......neg......ne9_ _............neg ............................neg.....................40.......
200706320.... .......ND.......>....Pos.......neg.......neg. .............Pos Pos 30.24 ......................................... ..:..
200706321 ND neg neg neg neg neg 40 .................................
...................................................................
.......................................
..........................................................
200706322 ND neg neg neg neg neg 40 200706323 ND neg neg neg neg neg 40 200706324 ND neg neg neg neg neg 40 200706325 ND neg neg..... neg...... neg neg ..... 40 200706326 ND neg neg neg neg neg 40 200706327 ND Pos neg neg Pos Pos 34.41 200706328 ND neg...... neg......neg, neg ne9...... 40 [0222] The entire genome sequence of this virus was completed using a classic primer walking sequencing approach on RNA. The complete genome of the Eb ebolavirus was not available, so it too was derived by a similar combination of random primed pyrosequencing and primer walking approaches. Acquisition of these sequences allowed for the first time the phylogenetic analysis of the complete genomes of representatives of all known species of Ebola and Marburg viruses. The analysis revealed that the newly discovered virus differed from the four existing ebolavirus species (Figure 1), with approximately 32% nucleotide difference from even the closest relative, EboIC
(Table 2). Similar complete genome divergence (35-45%) is seen between the previously characterized ebolavirus species.
[0223] Table 2. Identity matrix based on comparisons of full-length genome sequences of Zaire ebolaviruses 1976 (Genbank accession number NC_002549) and 1995 (Genbank accession number AY354458), Sudan ebolavirus 2000 (Genbank accession number NC_006432), Cote d'Ivoire ebolavirus 1994 (SEQ ID NO: 10), Reston ebolavirus 1989 (Genbank accession number NC_004161), and Bundibugyo ebolavirus 2007 (SEQ ID NO: 1).
Table 2 Zaire `95 Sudan `00 EboIC `94 EboBun `07 Reston `89 Zaire `76 .988 .577 .630 .632 .581 Zaire `95 .577 .631 .633 .581 Sudan `00 .577 .577 .609 EboIC `94 .683 .575 EboBun `07 .576 [0224] The material and information obtained from the discovery of the new unique virus EboBun and the realization that together with EboIC these viruses represent a Glade of Bundibungyo-Ivory Coast Ebola virus species is valuable, and makes possible the development of clinical, diagnostic and research tools directed to human hEbola infection.
Material and Methods [0225] Ebolavirus detection and virus isolation. Several diagnostic techniques were used for each sample: (i) antigen capture, IgG, and IgM assays were performed as previously described" (ii) virus isolation attempts were performed on Vero E6 cells12 and monitored for 14 days; (iii) RNA
was extracted and tested for Zaire16 and Sudan ebolavirus and marburgvirus4 using real-time quantitative RT-PCR assays designed to detect all known species of each respective virus species the primers/probe for the Sudan ebolavirus assay were EboSudBMG 1(+) 5'-GCC ATG
GIT TCA GGT
TTG AG-3' (SEQ ID NO: 21), EboSudBMG 1(-) 5'-GGT IAC ATT GGG CAA CAA TTC A-3' (SEQ ID NO: 22) and Ebola Sudan BMG Probe 5'FAM-AC GGT GCA CAT TCT CCT TTT CTC
GGA-BHQ1 (SEQ ID NO: 23)]; (iv) the conventional RT-PCR was performed with the filo A/B
primer set as previously described16 using Superscript III (Invitrogen) according to the manufacturer's instructions. The specimen 200706291 was selected as the reference sample for further sequence analysis.
[0226] Genome sequencing. Pyrosequencing was carried out utilizing the approach developed by 454 Life Sciences, and the method described by Cox-Foster et al. 8 Subsequent virus whole genome primer walking was performed as previously described17 but using the primers specific for Bundibugyo ebolavirus RT-PCR amplification. In total, the entire virus genome was amplified in six overlapping RT-PCR fragments (all primers listed 5' to 3'): fragment A
(predicted size 2.7 kb) was amplified using forward-GTGAGACAAAGAATCATTCCTG (SEQ ID NO: 24) with reverse-CATCAATTGCTCAGAGATCCACC (SEQ ID NO: 25); fragment B (predicted size 3.0 kb) was amplified using forward-CCAACAACACTGCATGTAAGT (SEQ ID NO: 26) with reverse-AGGTCGCGTTAATCTTCATC (SEQ ID NO: 27); fragment C (predicted size 3.5 kb) was amplified using forward-GATGGTTGAGTTACTTTCCGG (SEQ ID NO: 28) with reverse-GTCTTGAGTCATCAATGCCC (SEQ ID NO: 29); fragment D (predicted size 3.1 kb) was amplified using forward-CCACCAGCACCAAAGGAC (SEQ ID NO: 30) with reverse-CTATCGGCAATGTAACTATTGG (SEQ ID NO: 31); fragment E (predicted size 3.4 kb) was amplified using forward-GCCGTTGTAGAGGACACAC (SEQ ID NO: 32) with reverse-CACATTAAATTGTTCTAACATGCAAG (SEQ ID NO: 33) and fragment F (predicted size 3.5 kb) was amplified using forward-CCTAGGTTATTTAGAAGGGACTA (SEQ ID NO: 34) with reverse-GGT AGA TGT ATT GAC AGC AAT ATC (SEQ ID NO: 35).
[0227] The exact 5' and 3' ends of Bundibugyo ebolavirus were determined by 3' RACE from virus RNA extracted from virus infected Vero E6 cell monolayers using TriPure isolation reagent.
RNAs were then polyadenylated in vitro using A-Plus poly(A) polymerase tailing kit (Epicenter Biotechnologies) following the manufacturer's instructions and then purified using an RNeasy kit (Qiagen) following standard protocols. Ten microliters of in vitro polyadenylated RNA were added as template in RT--PCR reactions, using SuperScript III One--Step R'I'-PCR
system with Platinum Tack High Fidelity (Invitrogen) following the manufacturer's protocol. Two parallel RT-PCR
reactions using the oligo(dT)-containing 3'RACf-AP primer (Invitrogen) mixed with I of 2 viral specific primers, Ebo-U 692(-) ACAAAAAGCTATCTGCACTAT (SEQ ID NO: 36) and Ebo-U18269(+) CTCAGAAGCAAAATTAATGG (SEQ ID NO: 37), generated -700 nt long fragments containing the 3' ends of either genomic and antigenomic RNAs. The resulting RT-PCR products were analyzed by agarose electrophoresis, and DNA bands of the correct sizes were purified using QlAquick Gel Extraction Kit (Qiagen) and sequenced using standard protocols (ABI).
[0228] The nucleotide sequence of the Cote d'Ivoire ebolavirus (EboIC) isolate RNA was initially determined using the exact same pyrosequencing strategy as that used for Bundibugyo ebolavirus described above. This method generated sequence for approximately 70% of the entire genome. This draft sequence was then used to design a whole genome primer walking strategy for filling any gaps and confirming the initial sequence. The following Cote d'Ivoire ebolavirus-specific primers were used to generate RT-PCR fragments, designated A-F, as follows: Fragment A
(predicted size 3.0 kb) was amplified using forward-GTGTGCGAATAACTATGAGGAAG
(SEQ
ID NO: 38) and reverse-GTCTGTGCAATGTTGATGAAGG (SEQ ID NO: 39); Fragment B
(predicted size 3.2 kb) was amplified using forward-CATGAAAACCACACTCAACAAC
(SEQ ID
NO: 40) and reverse-GTTGCCTTAATCTTCATCAAGTTC (SEQ ID NO: 41); Fragment C
(predicted size 3.0 kb) was amplified using forward-GGCTATAATGAATTTCCTCCAG
(SEQ ID
NO: 42) and reverse-CAAGTGTATTTGTGGTCCTAGC (SEQ ID NO: 43); fragment D
(predicted size 3.5 kb) was amplified using forward-GCTGGAATAGGAATCACAGG (SEQ ID NO: 44) and reverse-CGGTAGTCTACAGTTCTTTAG (SEQ ID NO: 45); fragment E (predicted size 4.0 kb) was amplified using forward-GACAAAGAGATTAGATTAGCTATAG (SEQ ID NO: 46) and reverse-GTAATGAGAAGGTGTCATTTGG (SEQ ID NO: 47); fragment F (predicted size 2.9 kb) was amplified using forward-CACGACTTAGTTGGACAATTGG (SEQ ID NO: 48) and reverse-CAGACACTAATTAGATCTGGAAG (SEQ ID NO: 49); fragment G (predicted size 1.3 kb) was amplified using forward-CGGACACACAAAAAGAAWRAA (SEQ ID NO: 50) and reverse-CGTTCTTGACCTTAGCAGTTC (SEQ ID NO: 51); and fragment H (predicted size 2.5 kb) was amplified using forward-GCACTATAAGCTCGATGAAGTC (SEQ ID NO: 52) and reverse-TGGACACACAAAAARGARAA (SEQ ID NO: 53). A gap in the sequence contig was located between fragments C and D and this was resolved using the following primers to generate a predicted fragment of 1.5 kb: forward-CTGAGAGGATCCAGAAGAAAG (SEQ ID NO: 54) and reverse-GTGTAAGCGTTGATATACCTCC (SEQ ID NO: 55). The terminal -20 nucleotides of the sequence were not experimentally determined but were inferred by comparing with the other known Ebola genome sequences.
[0229] Bundibugyo ebolavirus real-time RT-PCR assay. The primers and probe used in the Bundibugyo ebolavirus specific Q-RT-PCR assay were as follows: EboU965( +): 5'-GAGAAAAGGCCTGTCTGGAGAA-3' (SEQ ID NO: 56), EboU1039(-): 5'-TCGGGTATTGAATCAGACCTTGTT-3' (SEQ ID NO: 57) and EboU989 Prb: 5'Fam-TTCAACGACAAATCCAAGTGCACGCA-3'BHQ1 (SEQ ID NO 58). Q-RT-PCR reactions were set up using Superscript III One-Step Q-RT-PCR (Invitrogen) according to the manufacturer's instructions and run for 40 cycles with a 58 C annealing temperature.
[0230] Phylogenetic analysis. Modeltest 3.718 was used to examine 56 models of nucleotide substitution to determine the model most appropriate for the data. The General Time Reversible model incorporating invariant sites and a gamma distribution (GTR+I+G) was selected using the Akaike Information Criterion (AIC). Nucleotide frequencies were A = 0.3278, C
= 0.2101, G =
0.1832, T = 0.2789, the proportion of invariant sites = 0.1412, and the gamma shape parameter =
1.0593. A maximum likelihood analysis was subsequently performed in PAUP*4.ObIO19 using the GTR+I+G model parameters. Bootstrap support values were used to assess topological support and were calculated based on 1,000 pseudoreplicates20.
[0231] In addition, a Bayesian phylogenetic analysis was conducted in MrBayes 3.221 using the 5 GTR+I+G model of nucleotide substitution. Two simultaneous analyses, each with four Markov chains, were run for 5,000,000 generations sampling every 100 generations.
Prior to termination of the run, the AWTY module was used to assess Markov Chain Monte Carlo convergence to ensure that the length of the analysis was sufficient22. Trees generated before the stabilization of the likelihood scores were discarded (burn in = 40), and the remaining trees were used to construct a 10 consensus tree. Nodal support was assessed by posterior probability values (> 95 = statistical support).
Example 2 [0232] Immunization against EboBun:
15 [0233] To determine the capability of immunogens to elict an immune response in non-human primates (NHP), 12 cynomolgus macaques, of which 10 are immunized with VSVAG/EboBunGP
either orally (OR; n = 4), intranasally (IN; n = 4) or intramuscularly (IM; n = 2) in accordance with all animal control and safety guidelines and essentially as described by Qiu, X, et al., PLoS ONE. 2009;
4(5): e5547. The remaining 2 control animals are vaccinated intramuscularly with 20 VSVAG/MARVGP. VSVAG/MARVGP does not provide heterologous protection against EboBun, therefore these NHPs succumb to EboBun infection. Animals are acclimatized for 14 days prior to infection. Animals are fed and monitored twice daily (pre- and post-infection) and fed commercial monkey chow, treats and fruit. Husbandry enrichment consists of commercial toys and visual stimulation.
25 [0234] The recombinant VSVAG/EboBun vaccines are synthesized expressing the EboBun glycoprotein (GP) (SEQ ID NO: 9), soluble glycoprotein (sGP) (SEQ ID NO: 4), or nucleoprotein (NP) (SEQ ID NO: 3). Control VSVAG/MARVGP vaccines represent the analogous proteins from Lake victoria marburgvirus (MARV) (strain Musoke). The following results for GP are similar for sGP and NP. Vaccines are generated using VSV (Indiana serotype) as described previously.
30 Garbutt, M, et al., J Virol, 2004; 78(10):5458-5465; Schnell, MJ, et al., PNAS USA, 1996;
93(21):11359-11365. EboBun challenge virus is passaged in Vero E6 cells prior to challenge, as described previously Jones, SM, et al., Nat Med, 2005; 11(7):786-790;
Jahrling, PB, et al., J Infect Dis, 1999; 179(Suppl 1):S224-34. An EboBun immunogen peptide pool consisting of 15mers with 11 amino acid overlaps (Sigma-Genosys) spanning the entire sequence of the EboBun immunogens and strain Mayinga 1976 GP are used.
[0235] Twelve filovirus naive cynomolgus monkeys randomized into four groups receive 2 ml of lx107 PFU/ml of vaccine in Dulbecco's modified Eagle's medium (DMEM).
Animals in the three experimental groups are vaccinated with either: 1) 2 ml orally (OR) (n = 4);
2) 1 ml dripped into each nostril, intranasally (IN) (n = 4); or 3) 1 ml each into two sites intramuscularly (IM) (n = 2).
The two controls are injected intramuscularly with 2 ml of lx107 PFU/ml of VSVAG/MARVGP. All animals are challenged intramuscularly 28 days later with 1,000 PFU of EboBun.
[0236] Routine examination is conducted on 0, 2, 4, 6, 10, 14 and 21 days post-vaccination, then 0, 3, 6, 10, 14, 19, 26 days, 6 and 9 months after the EboBun challenge.
For the examinations animals are anaesthetized by intramuscular injection with 10 mg/kg of ketaset (Ayerst).
Examinations include haematological analysis, monitoring temperature (rectal), respiration rate, lymph nodes, weight, hydration, discharges and mucous membranes. Also, swabs (throat, oral, nasal, rectal, vaginal) and blood samples are collected (4 ml from femoral vein, 1 ml in EDTA vacutainer tube; 3 ml in serum separator vacutainer tube). Cynomolgus monkey PBMCs are isolated using BD
CPT sodium citrate Vacutainers (Becton Dickinson) as per manufacturer's protocol.
[0237] All VSVAG/EboBunGP immunized animals are protected from high dose challenge.
These animals show no evidence of clinical illness after vaccination or EboBun challenge. Both control animals demonstrate typical symptoms associated with EboBun HF
including fever, macular rashes, lethargy, and unresponsiveness. Continued infection requires euthanization. Hematology analyses at each examination date demonstrate increases in the platelet-crit in the OR and IN groups post-challenge, however, no significant changes are observed in any NHPs post-immunization or in the VSVAG/EboBunGP immunized NHPs post-challenge.
[0238] EboBun antibody production from humoral antibody response to vaccination and challenge is examined by a virus like particle (VLP) based ELISA assay.
Generation of EboBun VLPs is performed by the protocol for ZEBOV as described by Wahl-Jensen, V., et al., J Virol, 2005; 79(4):2413-2419. ELISA is performed by the protocol described by Qiu, X, et al., PLoS ONE.
2009; 4(5): e5547.
[0239] The VSVAG/MARVGP immunized animals do not develop a detectable antibody response to EboBun. In contrast, potent antibody responses are detected in all VSVAG/EboBunGP
immunized animals independent of immunization route. Between days 14 and 21 post-vaccination, all VSVAG/EboBunGP immunized NHPs develop high levels of IgA, IgM, and IgG
against EboBunGP. After challenge the IgM titres do not exceed the post-vaccination levels, however, IgG
and IgA antibody titres are increased peaking 14 days post-challenge then slowly decreasing before maintaining a relatively high antibody titre up to 9 months.
[0240] The level of neutralization antibodies is detected by a EboBun-GFP flow cytometric neutralization assay in serum collected at days 0 and 21 post-vaccination.
Samples are assayed in duplicate for their ability to neutralize an infection with EboBun-GFP in VeroE6 cells. Serially diluted serum samples are incubated with an equal volume of EboBun-GFP in DMEM, at 37 C, 5%
CO2 for 1 hr followed by addition of 150 l per well of a confluent 12 well plate of VeroE6 cells (MOI = 0.0005). After 2 hours at 37 C, 5% C02, 1 ml of DMEM, 2% fetal bovine serym (FBS), 100 U/ml penicillin, 100 g/ml streptomycin is added per well and incubated for 5 days. Cells are harvested by removing the culture supernatant, washing with 1 ml PBS, 0.04%
EDTA, then adding 800 l of PBS 0.04% EDTA for 5 minutes at 37 C before adding 8 ml PBS, 4%
paraformaldehyde (PFA) and overnight incubation. The cells are acquired (10,000 events) and analyzed with CellQuest Pro v3.3 on a Becton Dickinson FACSCalibur flow cytometer.
[0241] The OR and IN routes produce EboBunGP-specific neutralizing antibodies with the OR
route producing the highest titres post-vaccination. The IM immunization produces detectable levels of neutralizing antibody. In comparison, 3/4 NHPs in the OR group demonstrate a 50% reduction in EboBun-GFP positive cells at a titre of 1:40. Similarly, the IN route results in a reduction of EboBun-GFP positive cells at the 1:40 dilution.
[0242] EboBunGP-specific effector cellular immune responses are determined using IL-2 and IFN-y ELISPOT assays as described by Qin, X, et al., PLoS ONE. 2009; 4(5):
e5547 to determine the number of IL-2 and IFN-y secreting lymphocytes. Prior to challenge on days 10 to 14 post-vaccination there is a detectable EboBun immunogen-specific IFN-y response in all immunized animals. The IM route is the most potent, inducing approximately 2-fold more IFN-y secreting cells than OR (p<0.001) or IN (p = 0.043) routes. A strong post-challenge secondary IFN-y response is induced in all VSVAG/EboBun immunized animals with the IM route producing the most IFN-y cells at day 6. By day 10 the OR group demonstrates a stronger response. The IFN-y in the IN
group rises steadily, peaking at day 26 post-challenge with 4.3 and 2 fold more EboBun specific IFN-y secreting cells than the IM (p = 0.003) and OR (p = 0.075) group, respectively. All three routes produce strong EboBun-specific IFN-y responses.
[0243] Post-vaccination, the IM group also has more EboBunGP-specific IL-2 secreting cells than either of the mucosally immunized groups. Post-challenge, the IM route continues to dominate early after challenge peaking on day 10. This difference shows a trend when compared to the IN
group (p = 0.067) and is significant when compared to the OR group (p<0.001).
Additionally, the IN
group has more IL-2 producing cells than the OR group (p = 0.090) on day 10 post-challenge. By day 26 post-challenge all three routes continue to produce a EboBunGP-specific IL-2 response, however, the IN group response is strongest. At day 26 post-challenge the IN
group has the most potent IFN-y and IL-2 responses, as well as the highest IgA and IgG antibody titre, indicating this immunization route, followed by a EboBun challenge, results in the development of potent and sustained effector responses.
[0244] Absolute lymphocyte numbers for CD3+, CD4+, and CD8+ (CD3+4-) T cell populations are determined by flow cytometry. No decrease is observed in the lymphocyte populations for any of the VSVAG/EboBunGP vaccinated NHPs. In contrast, control animals who are not protected from EboBun show lymphocyte numbers decreased by 28-57%.
[0245] Macrophage numbers are slightly increased in control animals. However, the number of CD14+ cells is greater in the VSVAG/EboBunGP vaccinated groups with the IM
route showing the most significant increases.
[0246] In order to determine the long term immune response after challenge, EboBunGP-specific CD4+ and CD8+ memory T-lymphocytes are examined for their ability to proliferate (CFSE-) or produce IFN-y in response to EboBunGP peptides at 6 months post-vaccination.
EboBunGP-specific memory responses are observed as a result of vaccination followed by a ZEBOV challenge. These responses persist for at least 6 months. The memory populations in OR
and IN inoculation routes demonstrate the greatest potential for proliferation and IFN-y production post-challenge.
[0247] Any patents or publications mentioned in this specification are incorporated herein by reference to the same extent as if each individual publication is specifically and individually indicated to be incorporated by reference.
[0248] The compositions and methods described herein are presently representative of preferred embodiments, exemplary, and not intended as limitations on the scope of the invention. Changes therein and other uses will occur to those skilled in the art. Such changes and other uses can be made without departing from the scope of the invention as set forth in the claims. All numerical ranges are inclusive of the whole integers and decimals between the endpoints, and inclusive of the endpoints.
References 1. Suzuki, Y., and Gojobori, T., (1997) The origin and evolution of Ebola and Marburg viruses. Mol Bio Evol, 14(8): 800-806.
2. Sanchez, A., Geisbert, T.W., Feldmann, H. in Fields Virology (ed. Knipe, D.
M., Howley, P.M.) 1409 - 1448 (Lippincott Williams and Wilkins, Philadelphia, 2007).
310 Leroy, E.M. et al., (2005) Fruit bats as reservoirs of Ebola virus.
Nature, 438, 575-6.
4. Towner, J.S. et al., (2007) Marburg virus infection detected in a common African bat. PLoS ONE, 2(8), e764.
5. Swanepoel, R. et al., (2007) Studies of reservoir hosts for Marburg virus.
Emerg Infect Dis, 13(12), 1847-51.
615 Le Guenno, B. et al., (1995) Isolation and partial characterization of a new species of Ebola virus.
Lancet, 345(8960), 1271-4.
7. Ksiazek, T. G. et al. (1999) Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, IgG and IgM antibody findings among EHF patients in Kikwit, 1995. J.
Infect Dis 179 (suppl 1), S177-S187.
820 Cox-Foster, D. L. et al. (2007) A metagenomic survey of microbes in honey bee colony collapse disorder. Science 318, 283-7.
9. World Health Organization (2008) Ebola outbreak contained in Uganda.
Features, 22 February, www.who.int/features/2008/ebola-outbreak/en/.
10. Sullivan, N. J., Sanchez, A., Rollin, P. E., Yang, Z.-Y. & Nabel, G. J.
(2000) Development of a 25 preventive vaccine for Ebola virus infection in primates. Nature 408, 605-609.
11. Ksiazek, T. G., West, C. P., Rollin, P. E., Jahrling, P. B. & Peters, C.
J. (1999) ELISA for the detection of antibodies to Ebola viruses. J. Infect Dis 179 (suppl 1), S191-S198.
12. Rodriguez, L. et al. (1999) Persistence and genetic stability of Ebola virus during the outbreak in Kikwit, Zaire 1995. J. Infect Dis 179 (suppl 1), S170-S176.
130 Sanchez, A. et al. Detection and molecular characterization of Ebola viruses causing disease in human and nonhuman primates. J. Infect Dis 179 (suppl 1), S164-S169 (1999).
14. Jones, S. M. et al. (2005) Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat Med 11, 786-90.
15. Geisbert, T. W. et al. (2008) Recombinant vesicular stomatitis virus vector mediates postexposure protection against Sudan Ebola hemorrhagic fever in nonhuman primates. J Virol 82, 5664-8.
165 Towner, J. S., Sealy, T. K., Ksiazek, T. & Nichol, S. T. (2007) High-throughput molecular detection of hemorrhagic fever virus threats with applications for outbreak settings. J.
Inf Dis 196 (suppl 2), S205-212.
17. Towner, J. S. et al. (2006) Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J Virol 80, 6497-516.
1$0 Posada, D. & Crandall, K. A. (1998) MODELTEST: testing the model of DNA
substitution.
Bioinformatics 14, 817-818.
19. Swofford, D. L. (2002) PAUP*: phylogenetic analysis using parsimony (*and other methods) version 4.0blO. Sinauer Assoc., Sunderland, Mass.
20. Felsenstein, J. (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 15 39, 783-791.
21. Ronquist, F. & Huelsenbeck, J. P. (2003) MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572-1574.
22. Nylander, J. A. A., Wilgenbusch, J. C., Warren, D. L. & Swofford, D. L.
(2008) AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics.
20 Bioinformatics 24, 581-583.
LUOKITTELU
| Kansainvälinen luokitus | A61K39/12, C07K14/08, C07K16/10, C12N7/01, C12N15/40, G01N33/68 |
| Yhteistyöluokitus | C07K16/10, C12N2760/14145, C07K2316/96, A61K2039/5256, C12N2810/6072, A61K2039/53, A61K39/12, C12N2760/14143,G01N2333/08, G01N33/56983, C12N7/00 |
| Eurooppalainen luokitus | C07K16/10, C12N7/00, G01N33/569K, A61K39/12 |
OIKEUDELLISET TAPAHTUMAT
| Päiväys | Koodi | Tapahtuma | Kuvaus |
|---|---|---|---|
| 21. heinäkuu 2014 | EEER | Examination request |
Effective date: 20140714
|
Ebola, AIDS Manufactured By Western Pharmaceuticals, US DoD?
Tue, 09/09/2014 - 09:59 admin
Scientists Allege
By:
Dr. Cyril Broderick, Professor of Plant Pathology
Dear World Citizens:
I have read a number of articles from your Internet outreach as well as articles from other sources about the casualties in Liberia and other West African countries about the human devastation caused by the Ebola virus. About a week ago, I read an article published in the Internet news summary publication of the Friends of Liberia that said that there was an agreement that the initiation of the Ebola outbreak in West Africa was due to the contact of a two-year old child with bats that had flown in from the Congo. That report made me disconcerted with the reporting about Ebola, and it stimulated a response to the “Friends of Liberia,” saying that African people are not ignorant and gullible, as is being implicated. A response from Dr. Verlon Stone said that the article was not theirs, and that “Friends of Liberia” was simply providing a service. He then asked if he could publish my letter in their Internet forum. I gave my permission, but I have not seen it published. Because of the widespread loss of life, fear, physiological trauma, and despair among Liberians and other West African citizens, it is incumbent that I make a contribution to the resolution of this devastating situation, which may continue to recur, if it is not properly and adequately confronted. I will address the situation in five (5) points:
1. EBOLA IS A GENETICALLY MODIFIED ORGANISM (GMO)
Horowitz (1998) was deliberate and unambiguous when he explained the threat of new diseases in his text, Emerging Viruses: AIDS and Ebola - Nature, Accident or Intentional. In his interview with Dr. Robert Strecker in Chapter 7, the discussion, in the early 1970s, made it obvious that the war was between countries that hosted the KGB and the CIA, and the ‘manufacture’ of ‘AIDS-Like Viruses’ was clearly directed at the other. In passing during the Interview, mention was made of Fort Detrick, “the Ebola Building,” and ‘a lot of problems with strange illnesses’ in “Frederick [Maryland].” By Chapter 12 in his text, he had confirmed the existence of an American Military-Medical-Industry that conducts biological weapons tests under the guise of administering vaccinations to control diseases and improve the health of “black Africans overseas.” The book is an excellent text, and all leaders plus anyone who has interest in science, health, people, and intrigue should study it. I am amazed that African leaders are making no acknowledgements or reference to these documents.
2. EBOLA HAS A TERRIBLE HISTORY, AND TESTING HAS BEEN SECRETLY TAKING PLACE IN AFRICA
I am now reading The Hot Zone, a novel, by Richard Preston (copyrighted 1989 and 1994); it is heart-rending. The prolific and prominent writer, Steven King, is quoted as saying that the book is “One of the most horrifying things I have ever read. What a remarkable piece of work.” As a New York Times bestseller, The Hot Zone is presented as “A terrifying true story.” Terrifying, yes, because the pathological description of what was found in animals killed by the Ebola virus is what the virus has been doing to citizens of Guinea, Sierra Leone and Liberia in its most recent outbreak: Ebola virus destroys peoples’ internal organs and the body deteriorates rapidly after death. It softens and the tissues turn into jelly, even if it is refrigerated to keep it cold. Spontaneous liquefaction is what happens to the body of people killed by the Ebola virus! The author noted in Point 1, Dr. Horowitz, chides The Hot Zone for writing to be politically correct; I understand because his book makes every effort to be very factual. The 1976 Ebola incident in Zaire, during President Mobutu Sese Seko, was the introduction of the GMO Ebola to Africa.
3. SITES AROUND AFRICA, AND IN WEST AFRICA, HAVE OVER THE YEARS BEEN SET UP FOR TESTING EMERGING DISEASES, ESPECIALLY EBOLA
The World Health Organization (WHO) and several other UN Agencies have been implicated in selecting and enticing African countries to participate in the testing events, promoting vaccinations, but pursuing various testing regiments. The August 2, 2014 article, West Africa: What are US Biological Warfare Researchers Doing in the Ebola Zone? by Jon Rappoport of Global Research pinpoints the problem that is facing African governments.
Obvious in this and other reports are, among others:
(a) The US Army Medical Research Institute of Infectious Diseases (USAMRIID), a well-known centre for bio-war research, located at Fort Detrick, Maryland;
(b) Tulane University, in New Orleans, USA, winner of research grants, including a grant of more than $7 million the National Institute of Health (NIH) to fund research with the Lassa viral hemorrhagic fever;
(c) the US Center for Disease Control (CDC);
(d) Doctors Without Borders (also known by its French name, Medicins Sans Frontiers);
(e) Tekmira, a Canadian pharmaceutical company;
(f) The UK’s GlaxoSmithKline; and
(g) the Kenema Government Hospital in Kenema, Sierra Leone.
Reports narrate stories of the US Department of Defense (DoD) funding Ebola trials on humans, trials which started just weeks before the Ebola outbreak in Guinea and Sierra Leone. The reports continue and state that the DoD gave a contract worth $140 million dollars to Tekmira, a Canadian pharmaceutical company, to conduct Ebola research. This research work involved injecting and infusing healthy humans with the deadly Ebola virus. Hence, the DoD is listed as a collaborator in a “First in Human” Ebola clinical trial (NCT02041715, which started in January 2014 shortly before an Ebola epidemic was declared in West Africa in March. Disturbingly, many reports also conclude that the US government has a viral fever bioterrorism research laboratory in Kenema, a town at the epicentre of the Ebola outbreak in West Africa. The only relevant positive and ethical olive-branch seen in all of my reading is that Theguardian.com reported, “The US government funding of Ebola trials on healthy humans comes amid warnings by top scientists in Harvard and Yale that such virus experiments risk triggering a worldwide pandemic.” That threat still persists.
4. THE NEED FOR LEGAL ACTION TO OBTAIN REDRESS FOR DAMAGES INCURRED DUE TO THE PERPETUATION OF INJUSTICE IN THE DEATH, INJURY AND TRAUMA IMPOSED ON LIBERIANS AND OTHER AFRICANS BY THE EBOLA AND OTHER DISEASE AGENTS.
The U. S., Canada, France, and the U. K. are all implicated in the detestable and devilish deeds that these Ebola tests are. There is the need to pursue criminal and civil redress for damages, and African countries and people should secure legal representation to seek damages from these countries, some corporations, and the United Nations. Evidence seems abundant against Tulane University, and suits should start there. Yoichi Shimatsu’s article, The Ebola Breakout Coincided with UN Vaccine Campaigns, as published on August 18, 2014, in the Liberty Beacon.
5. AFRICAN LEADERS AND AFRICAN COUNTRIES NEED TO TAKE THE LEAD IN DEFENDING BABIES, CHILDREN, AFRICAN WOMEN, AFRICAN MEN, AND THE ELDERLY. THESE CITIZENS DO NOT DESERVE TO BE USED AS GUINEA PIGS!
Africa must not relegate the Continent to become the locality for disposal and the deposition of hazardous chemicals, dangerous drugs, and chemical or biological agents of emerging diseases. There is urgent need for affirmative action in protecting the less affluent of poorer countries, especially African citizens, whose countries are not as scientifically and industrially endowed as the United States and most Western countries, sources of most viral or bacterial GMOs that are strategically designed as biological weapons. It is most disturbing that the U. S. Government has been operating a viral hemorrhagic fever bioterrorism research laboratory in Sierra Leone. Are there others? Wherever they exist, it is time to terminate them. If any other sites exist, it is advisable to follow the delayed but essential step: Sierra Leone closed the US bioweapons lab and stopped Tulane University for further testing.
The world must be alarmed. All Africans, Americans, Europeans, Middle Easterners, Asians, and people from every conclave on Earth should be astonished. African people, notably citizens more particularly of Liberia, Guinea and Sierra Leone are victimized and are dying every day. Listen to the people who distrust the hospitals, who cannot shake hands, hug their relatives and friends. Innocent people are dying, and they need our help. The countries are poor and cannot afford the whole lot of personal protection equipment (PPE) that the situation requires. The threat is real, and it is larger than a few African countries. The challenge is global, and we request assistance from everywhere, including China, Japan, Australia, India, Germany, Italy, and even kind-hearted people in the U.S., France, the U.K., Russia, Korea, Saudi Arabia, and anywhere else whose desire is to help. The situation is bleaker than we on the outside can imagine, and we must provide assistance however we can. To ensure a future that has less of this kind of drama, it is important that we now demand that our leaders and governments be honest, transparent, fair, and productively engaged. They must answer to the people. Please stand up to stop Ebola testing and the spread of this dastardly disease.
Thank you very much.
Sincerely,
Dr. Cyril E. Broderick, Sr.
About the Author:
Dr. Broderick is a former professor of Plant Pathology at the University of Liberia’s College of Agriculture and Forestry. He is also the former Observer Farmer in the 1980s. It was from this column in our newspaper, the Daily Observer, that Firestone spotted him and offered him the position of Director of Research in the late 1980s. In addition, he is a scientist, who has taught for many years at the Agricultural College of the University of Delaware.
Tässä alla on USA:n hallituksen kehittämä hiv-viruksen patentti, jolla USA on käynyt sotaan ihmiskuntaa vastaan. USA:n hallitus on pyrkinyt tappamaan tällä viruksella afrikkalaiset ja myös muut.
http://www.patents.com/us-4647773.html
United States Patent
4,647,773 Gallo , et al.March 3, 1987
Method of continuous production of retroviruses (HTLV-III) from patients with AIDS and pre-AIDS
Abstract
A cell system is disclosed for the reproducible detection and isolation of human T-lymphotropic retroviruses (HTLV-family) with cytopathic effects (HTLV-III) from patients with the acquired immune deficiency syndrome (AIDS), pre-AIDS and in healthy carriers. One neoplastic aneuploid T-cell line derived from an adult with lymphoid leukemia, and termed HT, was susceptible to infection with the new variants of HTLV, which are transformed and providing T-cell populations which are highly susceptible and permissive from HTLV-III, and convenience for large scale production, isolation and biological detection of the virus.
| Inventors: | Gallo; Robert C. (Bethesda, MD), Popovic; Mikulas (Bethesda, MD) |
| Assignee: | The United States of America as represented by the Department of Health(Washington, DC) |
| [*] Notice: | The portion of the term of this patent subsequent to May 28, 2002 has been disclaimed. |
| Appl. No.: | 06/602,946 |
| Filed: | April 23, 1984 |
| Current U.S. Class: | 435/239 ; 424/208.1; 435/235.1; 435/372.3; 435/948 |
| Current International Class: | C12N 5/06 (20060101); C12N 7/00 (20060101); C12N 007/02 (); C12N 007/00 (); C12N 005/00 (); C12R 001/91 () |
| Field of Search: | 435/235,239,240,948,5,29 424/89 128/1T |
| 4464465 | August 1984 | Lostrom et al. |
| 4520113 | May 1985 | Gallo et al. |
Popovic et al, Science, 224(4648):497-500, May 4, 1984. . Gallo et al, "Isolation of Human T-Cell Leukemia Virus in Acquired Immune Deficiency Syndrome (AIDS)", Science, 220, pp. 865-867 (5-1983). . Barre-Sinoussi et al, "Isolation of a T-Lymphotropic Retrovirus from a Patient at Risk for AIDS", Science, 220, pp. 868-871 (5-1983). . Marx, "Strong New Candidate for AIDS Agent", Science, 224, pp. 475-477 (5-1984).. |
Primary Examiner: Warren; Charles F.
Assistant Examiner: Tarcza; John Edward
Attorney, Agent or Firm: Roberts, Jr.; John S.
We claim:
1. A method for continuous production of HTLV-III virus which comprises infecting in cell culture highly susceptible, permissive cells consisting of a neoplastic aneuploid HT-cell line with said virus, said cells preserve the capacity for permanent growth after the infection with said virus, growing said cells under conditions suitable for cell growth and recovering said virus produced by said cells.
2. The method of claim 1, wherein said virus consists of cytopathic variants of HTLV.
3. The method of claim 1 wherein said infecting comprises cocultivating said virus with said cells to produce a cell line and recovering said cell line.
4. The method of claim 1 wherein said infecting comprises cell-free infection of said cells with said virus.
5. The method of claim 1 wherein said cells are neoplastic aneuploid T-cells derived from an adult with lymphoid leukemia.
6. A process for the continuous production of HTLV-III virus which comprises cocultivating said virus with a target HT-cell to produce an infected cell line, growing said cell line and recovering said virus from supernatants produced by said cell line.
7. The process of claim 6 wherein said target T-cell is a neoplastic aneuploid T-cell susceptible to infection with HTLV-III.
8. A process for the continual production of HTLV-III by infecting T-cells in cultures which comprises cocultivating HTLV-III virus with an HT-clone to produce an infected cell line, said clone being an aneuploid T-cell line derived from lymphoid leukemia, growing said cell line and recovering said virus from supernatants produced by said cell line.
9. The process in claim 8 wherein said clone is a mature T-cell phenotype of OKT3.sup.+ (62%), OKT4.sup.+ (39%) and OKT8.sup.-.
10. A method of producing a cell line containing an antigen of HTLV-III which comprises infecting an HT-cell line derived from lymphoid leukemia and susceptible to infection with HTLV-III, said cell line is capable of continuous large-scale production of HTLV-III, and growing the infected cell line under conditions suitable for growth.
11. The method of claim 10 wherein said cell line is a neoplastic aneuploid T-cell line.
12. The method of claim 10 wherein said HTLV-III are variants of human T-lymphotropic retrovirus, exhibit cytopathic effects and are non-transforming.
13. A cell line containing HTLV-III designated H9/HTLV-III.sub.B, ATCC Accession CRL 8543.
14. A process for producing a cell line H9/HTLV-III.sub.B which comprises infecting a target T-cell with HTLV-III virus to produce a cell line and recovering same, said infecting process overcomes the normal cytopathic effect of HTLV-III and preserves the immortal growth capacity of the target cell.
15. An HT cell line permanently infected with HTLV-III virus, wherein said cell line continually produces said virus.
The present invention describes a cell system for the reproducible detection and isolation of human T-lymphotropic retroviruses (HTLV-family) with cytopathic or cell killing effects (HTLV-III) from patients with the acquired immune deficiency syndrome (AIDS), pre-AIDS and in healthy carriers. One neoplastic aneuploid T-cell line derived from an adult with lymphoid leukemia, and termed HT, was susceptible to infection with the new variants of HTLV, providing T-cell populations which are highly susceptible and permissive for HTLV-III, and convenience for large scale production, isolation, and biological detection of the virus.
BACKGROUND OF THE INVENTION
The disclosure of this invention is contained in the following journal articles: Gallo et al., "Detection, Isolation, and Continuous Production of Cytopathic Human T-Lymphotropic Retroviruses (HTLV-III) from Patients with AIDS and pre-AIDS," Science, in press; and Gallo et al., "Human T-Lymphotropic Retrovirus, HTLV-III, Isolated from AIDS Patients and Donors at Risk for AIDS," in press.
Epidemiologic data strongly suggest that acquired immune deficiency syndrome (AIDS) is caused by an infectious agent which is apparently horizontally transmitted by intimate contact or blood products. Though the disease is manifested by opportunistic infections, predominantly Pneumocystis carcinii pneumonia and Kaposi's sarcoma, the underlying disorder affects the patient's cell-mediated immunity with absolute lymphopenia and reduced helper T-lymphocyte (OKT4.sup.+) subpopulation(s). Moreover, before a complete clinical manifestation of the disease occurs, its prodrome, pre-AIDS, is frequently characterized by unexplained chronical lymphadenopathy and/or leukopenia involving a helper T cell subset. This leads to the severe immune deficiency of the patient, suggesting that a specific subset of T-cells is the primary target for an infectious agent. Although patients with AIDS or pre-AIDS are often chronically infected with cytomegalovirus or hepatitis B virus, for various reasons these appear to be opportunistic or coincidental infections apparently not linked to the immunological response deficiency. It is believed that the cause of AIDS may be a virus from the family of human T-cell lymphotropic retroviruses (HTLV) which, prior to the present invention, comprised two major well characterized subgroups of human retroviruses, called human T-cell leukemia/lymphoma viruses, HTLV-I and HTLV-II. The most common isolate, HTLV-I, is mainly obtained from patients with mature T-cell malignancies. Seroepidemiological studies, in vitro biological effects, and nucleic acid hybridization data indicate that HTLV-I is etiologically associated with these malignancies, affecting adults primarily in the south of Japan, the Caribbean and Africa. HTLV of subgroup II (HTLV-II) was first isolated from a patient with a T-cell variant of hairy cell leukemia. To date, this is the only reported isolate of HTLV-II from a patient with a neoplastic disease. Virus isolation and seroepidemiological data show that HTLV of both subgroups can sometimes be found in patients with AIDS.
Evidence suggests that the retrovirus(es) of the HTLV family is an etiological agent of AIDS based on the following: (1) there is precedence for an animal retrovirus cause of immune deficiency (feline leukemia virus in cats); (2) retroviruses of the HTLV family are T-cell tropic; (3) they preferentially infect "helper" T-cells (OKT4.sup.+); (4) they have cytopathic effects on various human and mammalian cells as demonstrated by their induction of cell syncytia formation; (5) they can alter some T-cell functions; (6) in some cases infection may result in selective T-cell killing; and (7) they are transmitted by intimate contact or through blood products. The presence of antibodies directed to cell membrane antigens of HTLV infected cells has been shown in sera of more than 40% of patients with AIDS [Essex et al., Science, 220:859 (1983)]. This antigen has since been defined as part of the envelope of HTLV [Schupbach, et al., Science, in press; and Lee, et al., Proc. Nat. Acad. Sci. U.S.A., in press].
The original detection and isolation of the various HTLV isolates were made possible by two earlier developments: the discovery of T-cell growth factor (TCGF), also called Interleukin 2 (Il-2), which enabled the routine selective growth of different subsets of normal and neoplastic mature T-cells [Ruscetti, et al., J. Immunol., 119:131 (1977); and Poiesz, et al., Proc. Nat. Acad. Sci. U.S.A, 77:6134 (1980)] and the development of sensitive assays for detection of retroviruses based on reverse transcriptase assays. The methods of HTLV isolation and transmission involved a cocultivation procedure using permissive T-cells for the virus. The use of normal human T-cells in cocultivation experiments preferentially yielded HTLV of both subgroups with immortalizing (transforming) capability for some of the target T-cells.
However, HTLV variants (now termed HTLV-III), lack immortalizing properties for normal T-cells and mainly exhibit cytopathic effects on the T-cells and is now believed to be the cause of AIDS. In fact, such variants were frequently but only transiently detected using these normal T-cells as targets in cocultivation or cell-free transmission experiments. The cytopathic effect was overcome by finding a highly susceptible, permissive cell for cytopathic variants of HTLV, thus preserving the capacity for permanent growth after infection with the virus. The present invention discloses the identification and characterization of this new immortalized T-cell population and its use in the isolation and continuous high- level production of such viruses from patients with AIDS and pre-AIDS.
Early experiments identified one neoplastic aneuploid T-cell line, termed HT, derived from an adult with lymphoid leukemia, that was susceptible to infection with the new cytopathic virus isolates.
This cell line is a sensitive target for transmission of these virus isolates (HTLV-III) and it allows continuous large-scale virus production and development of specific immunologic reagents and nucleic acid probes useful for comparison of these new isolates among themselves and with HTLV-I and HTLV-II. In addition to their differences in biological effects that distinguish them from HTLV-I and HTLV-II, HTLV-III also differs from these known HTLV subgroups in several immunological assays and in morphology. However, these new retroviruses are T4 lymphotropic and exhibit many properties similar to HTLV-I and II, including similar properties of the reverse transcriptase, cross reactivity of structural proteins as determined by heterologous competition radioimmune assays with patients' sera and with animal hyperimmune sera, and induction of syncytia. These new retrovirus isolates are collectively designated HTLV-III, together with detectable differences in some of their proteins and genetic information, HTLV-III's ability to kill T-cells clearly separates these variants from other members of the HTLV family.
STATEMENT OF DEPOSIT
A cell line corresponding to the present invention, and denoted H9/HTLV-III.sub.B, has been deposited in the ATCC (under ATCC No. CRL 8543) on April 19, 1984, prior to the filing of this patent application. This deposit assures permanence of the deposit and ready accessibility thereto by the public. H9 is a representative and preferred cell line in accordance with the invention.
UTILITY STATEMENT
The cell line which is a product of the present invention (H9/HTLV-III.sub.B) is presently useful for the production of vaccines for the relief of AIDS and for the detection of antibodies to the virus in blood samples.
GENERAL DESCRIPTION
A susceptible cell line HT was tested for HTLV before in vitro infection and it was negative by all criteria, including lack of proviral sequences. Continuous production of HTLV-III is obtained after repeated exposure of parental HT cells (3.times.10.sup.6 cells pretreated with polybrene) to concentrated culture fluids containing HTLV-III harvested from short term cultured T-cells (grown with TCGF) which originated from patients with pre-AIDS or AIDS. The concentrated fluids were first shown to contain particle associated reverse transcriptase (RT). When cell proliferation declined, usually 10 to 20 days after exposure to the culture fluids, the fresh (uninfected) HT parental cells are added to cultures. Culture fluids from the infected parental cell line was positive for particulate RT activity and about 20% of the infected cell population was positive in an indirect immune fluorescent assay (IFA) using serum from a hemophilia patient with pre-AIDS (patient E.T.). Serum from E.T. also contained antibodies to proteins of disrupted HTLV-III but did not react with proteins of HTLV-I or HTLV-II infected cells.
SPECIFIC DISCLOSURE
As has been mentioned above, an aneuploid HT-cell line exhibited the desired prerequisites for the continuous propagation of HTLV-III. This cell line is a neoplastic aneuploid T-cell line derived from an adult patient with lymphoid leukemia, selected for its mature T-cell phenotype [OKT3.sup.+ (62%), OKT.sup.4 + (39%) and OKT8.sup.- ], as determined by cytofluorometry using a fluorescence-activated cell sorter. Cultures of these cells are routinely maintained in RPMI/1640 with 20% fetal calf serum and antibiotics. These cultures are shown in Example 1, Table 1. Clone H9 is preferred, with Clone H4 being secondarily preferred.
HTLV-III culture fluids are isolated from cultured cells of patients with acquired immune deficiency syndrome (AIDS). Peripheral blood leukocytes from these patients are banded in Ficoll-Hypaque, incubated in growth media (RPMI 1640, 20% fetal bovine serum 0.29 mg/ml glutamine) containing 5 .mu.g/ml phytohemagglutinin (PHA-P) for 48 hours, at 37.degree. C. in a 5% CO.sub.2 atmosphere. The leukocytes are then refed with growth medium containing 10% purified T cell growth factor (TCGF); optionally, some of the cells also received rabbit antibody to alpha interferon. Cells and growth media from these lymphocytes are then assayed for the presence of HTLV subgroups I-III. Samples exhibiting more than one of the following were considered positive: repeated detection of a Mg.sup.++ dependent reverse transcriptase activity in supernatant fluids; virus observed by electron microscopy; intracellular expression of virus-related antigens detected with antibodies from sero-positive donors or with hyperimmune serum; or transmission of particles, detected by reverse transcriptase assays or by electron microscopic observation, to fresh human cord blood, bone marrow, or peripheral blood T-lymphocytes. All isolates not classified as either HTLV-I or HTLV-II by immunological or nucleic acid analysis were classified as HTLV-III. The cells in the HTLV-III producing cell cultures, characterized using established immunological procedures, are predominantly T-lymphocytes (E rosette receptor, OKT/3 and Leu/1 positive, with a T4 phenotype (OKT4, leu 3a positive). This process is also described by Gallo, et al., in Science, 220:865-867 (1983).
The infection of parental HT cells as well as other cloned cell populations occurs by exposure of these cells to concentrated or nonconcentrated culture fluids (cell-free infection) from T-cell cultures from AIDS or pre-AIDS patients, or by cocultivation; that is, HT cells are infected by exposure to HTLV-III containing cultures. The usual cell-free infection procedure is as follows: 2 to 5.times.10.sup.6 cells are treated with polybrene (2 .mu.g/ml) or DEAE dextran for 30 minutes in CO.sub.2 incubator at 37.degree. C., and then exposed to the virus inoculum (0.1 to 1 ml) for one hour in the incubator (CO.sub.2 /37.degree. C.). The cells are kept in suspension by shaking at regular intervals. After one hour of incubation a regular growth medium is added. The positivity of infected cultures for HTLV-III is assessed after one, two, and three weeks of cultivation.
The infection of HT cells (clones) is also obtained by cocultivation procedure--HT cells are mixed in various proportions (usually 1:5) with short- term cultured T-cells (about 5 to 20 days) from AIDS or pre-AIDS patients. The positivity for HTLV-III was scored by the detection of viral antigens or viral nucleic acid sequences in the infected recipient cells at various intervals (7, 14, 21 days, etc.) after cocultivation. The mixed cultures are maintained in growth medium for several months.
EXAMPLE 1
As shown in Table 1 below, single cell HT clones were isolated as described by Popovic, et al., in Neoplasma, 18:257 (1971), and Bach, et al., Immunol. Rev., 54:5 (1981) from a long-term cultured aneuploid HT-cell line exhibiting mature T-cell phenotype (OKT3.sup.+ [62%], OKT4.sup.+ [39%] and OKT8.sup.-) as determined by cytofluorometry using a fluorescence-activated cell sorter. The cultures were routinely maintained in RPMI/1640 with 20% fetal calf serum and antibiotics. The terminal cell density of the parental cell culture, seeded at a concentration of 2.times.10.sup.5 cells/milliliter of culture media, was in the range 10.sup.6 -1.5.times.10.sup.6 cells/ml after 5 days of culture.
For detection of multinucleated cells, cell smears were prepared from cultures 6 and 14 days after infection and stained with Wright-Giemsa. Cells having more than 5 nuclei were considered as multi-nucleated cells. Cloned cells from uninfected cultures also contained some multi-nucleated giant cells as well; however, the arrangement of multiple nuclei in a characteristic ring formation was lacking and the number of these cells was much less (0.7% to 10%).
Immunofluorescence positive cells were washed with phosphate-buffered saline (PBS) and resuspended in the same buffer at concentration 10.sup.6 cells per milliliter. Approximately 50 .lambda. of cell suspension were spotted on slides, air dried, and fixed in acetone for 10 min. at room temperature. Slides were stored at -20.degree. C. until use. Twenty microliters of either hyperimmune rabbit antiserum to HTLV-III (diluted 1/2000 in PBS) or serum from the patient (E.T.) diluted 1/8 in PBS was applied to cells and incubated for 50 min. at 37.degree. C. The fluorescein-conjugated antiserum to rabbit or human immunogloblin G was diluted and applied to the fixed cells for 30 min. at room temperature. Slides then were washed extensively before microscopic examinations. The uninfected parental cell line as well as the clones were consistently negative in these assays.
To determine reverse transcriptase activity, virus particles were precipitated from cell-free supernatant as follows: 0.4 ml of 4M NaCl and 3.6 ml of 30% (wt/vol.) polyethylene glycol (Carbowax 6000) were added to 8 ml of harvested culture fluids and the suspension was placed on ice overnight. The suspension was centrifuged in a Sorvall RC-3 centrifuge at 2000 rpm at 4.degree. C. for 30 min. The precipitate was resuspended in 300 .mu.l at 50% (vol/vol) glycerol (25 mM Tris-HCl, pH 7.5/5 mM dithiothreitol/150 mM KCl/0.025% Triton X-100. Particles were disrupted by addition of 100 .mu.l of 0.9% Triton X-100/1.5M KCl. Reverse transcriptase (RT) assays were performed as described by Poiesz, et al., Proc Nat. Acad. Sci. U.S.A., 77:7415 (1980) and expressed in cpm per milliliter culture medium.
TABLE 1 __________________________________________________________________________ Response of Cloned T-Cell Populations to HTLV-III Infection Clones Characteristics H3 H4 H6 H9 H17 H31 H35 H38 __________________________________________________________________________ Total cell number (.times. 10.sup.6) 6 days after infection 1 1.5 1.5 0.3 0.4 0.3 0.5 1.8 14 days after infection 2.2 7.3 7.5 10.0 4.7 5.0 4.5 3.2 Multinucleated cells (%) 6 days after infection 24 42 32 7 13 14 30 45 14 days after infection 45 48 45 30 22 45 60 60 Immunofluorescence positive cells (%) 6 days after infection Rabbit antiserum to HTLV-III 55 56 32 32 39 21 10 87 Patient serum (E.T.) 56 29 21 ND ND ND ND 73 14 days after infection Rabbit antiserum to HTLV-III 50 74 60 97 71 40 20 80 Patient serum 45 47 56 78 61 43 22 89 Reverse transcriptase activity (.times. 10.sup.4 cpm/ml) 6 days after infection 2.4 1.8 2.1 4.1 2.6 1.4 1.7 2.5 14 days after infection 16.2 18.1 16.1 20.2 17.1 13.4 15.1 18.2 __________________________________________________________________________ ND = not done
EXAMPLE 2
As shown in Table 2 below, cocultivation with H4 recipient T-cell clone was performed with fresh mononuclear cells from peripheral blood of patients RF and SN, respectively. In the case of patients BK and LS cocultivation was performed with T-cells grown in the presence of exogenous TCGF (10% V/V) for 10 days. The ratio recipient/donor (patients') cells was 1:5. The mixed cultures were maintained in RPMI/1640 (20% FCS and antibiotics) in the absence of exogenous TCGF. Cell-free infection of H9 T-cell clone was performed using concentrated culture fluids harvested from T-cell cultures of the patient WT. The T-cell cultures were grown in the presence of exogenous TCGF for two weeks before the culture fluids were harvested and concentrated. Cells of H9 clones were pretreated with polybrene (2 .mu.g/ml) for 20 min. and 2.times.10.sup.6 cells were exposed for one hr. to 0.5 ml of 100-fold concentrated culture fluids positive for particulate RT activity.
HTLV-III virus expression in both cocultured and cell-free infected cell cultures were assayed approximately one month after in vitro cultivation. There was considerable fluctuation in HTLV-III expression (see Table 2). For details of both reverse transcriptase (RT) assays and indirect immunofluorescence assays (IFA) see Example 1.
TABLE 2 __________________________________________________________________________ Isolation of HTLV-III from Patients with Pre-AIDS and AIDS Virus Expression IFA with RT Activity Rabbit Serum Human Serum (ET) Patient Diagnosis Origin (.times. 10 cpm) (% Positive) (% Positive) EM __________________________________________________________________________ RF AIDS Haiti 0.25 80 33 ND (heterosexual) SN Hemophiliac U.S. 6.3 10 ND + (lymphadenopathy) BK AIDS U.S. 0.24 44 5 + (homosexual) LS AIDS U.S. 0.13 64 19 + (homosexual) WT Hemophiliac U.S. 3.2 69 ND ND (lymphadenopathy) __________________________________________________________________________ RT = reverse transcriptase IFA = immunofluorescence assays EM = electron microscopy ND = not done
EXAMPLE 3
To select for high permissiveness for HTLV-III and to preserve permanent growth and continuous production of virus, extensive cloning of the HT parental T-cell population was performed. A total of 51 single-cell clones was obtained by both capillary and limited dilution techniques using irradiated mononuclear cells from peripheral blood of a healthy donor as a feeder. The growth of these cell clones was compared after HTLV-III infection. A representative example of response to virus infection of 8 T-cell clones which are susceptible to and permissive for HTLV-III is shown in Table 1. In parallel experiments, 2.times.10.sup.6 cells of each T-cell clone were exposed to 0.1 ml of concentrated virus. Then cell growth and morphology, expression of cellular viral antigen(s), and RT activity in culture fluids were assessed 6 and 14 days after infection. Although all 8 clones were susceptible to and permissive for the virus, there were considerable differences in their ability to proliferate after infection. The cell number decreased by 10% to 90% from the initial cell count within 6 days after infection, and a high proportion of multinucleated (giant) cells were consistently found in all 8 infected clones. The percentage of T-cells positive for viral antigen(s) determined by immunofluorescent assays with serum from AIDS patient (E.T.) and with hyperimmune rabbit serum raised against the whole disrupted HTLV-III ranged from 10% to over 80%. Fourteen days after infection, the total cell number and the proportion of HTLV-III positive cells increased in all 8 clones. The virus positive cultures consistently showed round giant cells which contained numerous nuclei. These multinucleataed giant cells are similar to those induced by HTLV-I and HTLV-II except that the nuclei exhibit a characteristic ring formation. Electron microscopic examinations showed that the cells released considerable amounts of virus.
EXAMPLE 4
To determine whether HTLV-III is continuously produced by the infected T-cells in long-term cultures, both virus production and cell viability of the infected clone, H4, were followed for several months. Although the virus production fluctuated, culture fluids harvested from the H4/HTLV-III cell cultures at approximately 14-day intervals consistently exhibited particulate RT activity which has been followed for over 5 months. The viability of the cells ranged from 65% to 85% and doubling time of the cell population, which is called H4/HTLV-III, was approximately 30-40 hours. Thus, this permanently growing T-cell population can continuously produce HTLV-III.
The yield of virus produced by H4/HTLV-III cells was assessed by purification of concentrated culture fluids through a sucrose density gradient and assays of particulate RT activity in each fraction collected from the gradient. The highest RT activity was found at density 1.16 g/ml, similar to other retroviruses.
| USA on kehittänyt laboratorioissaan nämä kaksi ihmiskuntaa tappavaa virusta, mutta niitä on paljon enemmänkin kuin vain nämä kaksi. |
VastaaPoistaMY NAME IS MARIAM FROM SOUTH AFRICA...I SAW THIS COMMENT ON POSITIVE BLOGS AND I WILL LOVE TO TELL EVERYBODY HOW MY STATUS CHANGES TO NEGATIVE, AND AM NOW A LIVING WITNESS OF IT AND I THINK ITS A SHAME ON ME IF I DON'T SHARE THIS LOVELY STORY WITH OTHER PEOPLE INFECTED WITH THIS DEADLY VIRUS...,HIV HAS BEEN ONGOING IN MY FAMILY... I LOST BOTH PARENTS TO HIV,. AND IT IS SO MUCH PAIN IVE NOT BEEN ABLE TO GET OVER.. AS WE ALL KNOW MEDICALLY THERE IS NO SOLUTION TO IT..AND MEDICATION IS VERY EXPENSIVE..SO SOMEONE INTRODUCED ME TO A NATIVE MEDICAL PRACTITIONER IN AFRICA..I HAD A JOB THERE TO EXECUTE SO I TOOK TIME TO CHECK OUT ON HIM.I SHOWED HIM ALL MY TESTS AND RESULTS.. I WAS ALREADY DIAGNOSED WITH HIV AND IT WAS ALREADY TAKING ITS TOWL ON ME.. I HAD SPENT THOUSANDS OF DOLLARS SO I DECIDED TO TRY HIM OUT...I WAS ON HIS DOSAGE FOR 1 MONTHS. ALTHOUGH I DIDNT BELIEVE IN IT, I WAS JUST TRYING IT OUT OF FRUSTRATION... AND AFTER 2 WEEKS, I WENT FOR NEW TESTS... AND YOU WONT BELIEVE THAT 5 DIFFERENT DOCTORS CONFIRMED IT THAT AM NEGATIVE..IT WAS LIKE A DREAM,,I NEVER BELIEVE AIDS HAS CURE..AM NOW NEGATIVE,,AM A LIVING WITNESS..I DONT KNOW HOW TO THANK THIS MAN... I JUST WANT TO HELP OTHERS IN ANY WAY I CAN..HAVE JOINED MANY FORUMS AND HAVE POSTED THIS TESTIMONIES AND ALOT OF PEOPLE HAS MAIL AND CALLED THIS MAN ON PHONE AND AFTER 2 WEEKS THEY ALL CONFIRMED NEGATIVE..BBC NEWS TOOK IT LIVE AND EVERY.. HOPE HE HELPS YOU OUT.. EVERYBODY SAW IT AND ITS NOW OUT IN PAPERS AND MAGAZINES THAT THERE'S NATIVE CURE FOR HIV AND ALL WITH THE HELP OF THIS MAN,,HAVE TRIED MY OWN PARTS AND ALL LEFT WITH YOU,,IF YOU LIKE TAKE IT OR NOT..GOD KNOWS HAVE TRIED MY BEST.ABOUT 97 PEOPLE HAVE BEEN CONFIRMED NEGATIVE THROUGH ME..AND THEY SEND MAILS TO THANKS ME AFTER THEY HAVE BEEN CONFIRMED NEGATIVE,,THIS MAN IS REAL..DON'T MISS THIS CHANCE,,HIV IS A DEADLY VIRUS,,GET RID OF IT NOW..
case there is anyone who has similar problem and still
looking for a way out, and he those cast all kind of spell like ::
Love Spells
Luck, Money Spells
Health, Well Being
Protection, Healing
Curses, ex, Breakups
NEW! Combo Spells
High Priestess Spells
Vampire Spells
Authentic Voodoo Spells
Custom, Other Spells
Business spells
Health/Healing spells
Cancer healing
Curse removal
Job spells
Healing from all kind of diseases
Love binding
Barrenness(need a child)
Need love
Lottery Spells
Promotions
Success
Money rituals
winning court case
Divorce spells
Low sperm count
Infertility in women
Breast enlargement/reduction
Penis enlargement/reduction
YOU CAN CONTACT HIM HERE AS (dr.abalaka@outlook.com) and also his state based number text him here if you're in the US: 760-935-3804 if you need any question contact me via here as mariambaurice@gmail.com i wish you best of luck and good health.